- 医療関係者向けホーム
- 循環器領域
- エパデールEM
- Clinical Study:国内第Ⅲ相検証試験〔非劣性検証、優越性検証〕 試験概要
 Clinical Study国内第Ⅲ相検証試験〔非劣性検証、優越性検証〕
Clinical Study国内第Ⅲ相検証試験〔非劣性検証、優越性検証〕
試験概要「国内第Ⅲ相検証試験〔非劣性検証、優越性検証〕」
- 持田製薬社内資料:国内第Ⅲ相検証試験(MND2119H31試験)<2022年6月20日承認、CTD 2.7.6.4>[承認時評価資料]
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
目的
高脂血症(TG高値)患者を対象に、投与12週後※1における投与前値※2からの血清TG値の変化率を指標として、エパデールEM 2g/日のエパデールカプセル1.8g/日に対する非劣性の検証およびエパデールEM 4g/日のエパデールEM 2g/日に対する優越性の検証を行うとともに、安全性を検討する。なお、エパデールEM 4g/日はエパデールカプセル2.7g/日より高曝露と推定され、エパデールEM 4g/日の安全性がエパデールカプセル2.7g/日に劣らないことを、直接比較可能なデータに基づき示す必要があったため、エパデールカプセル2.7g/日群を設定した。
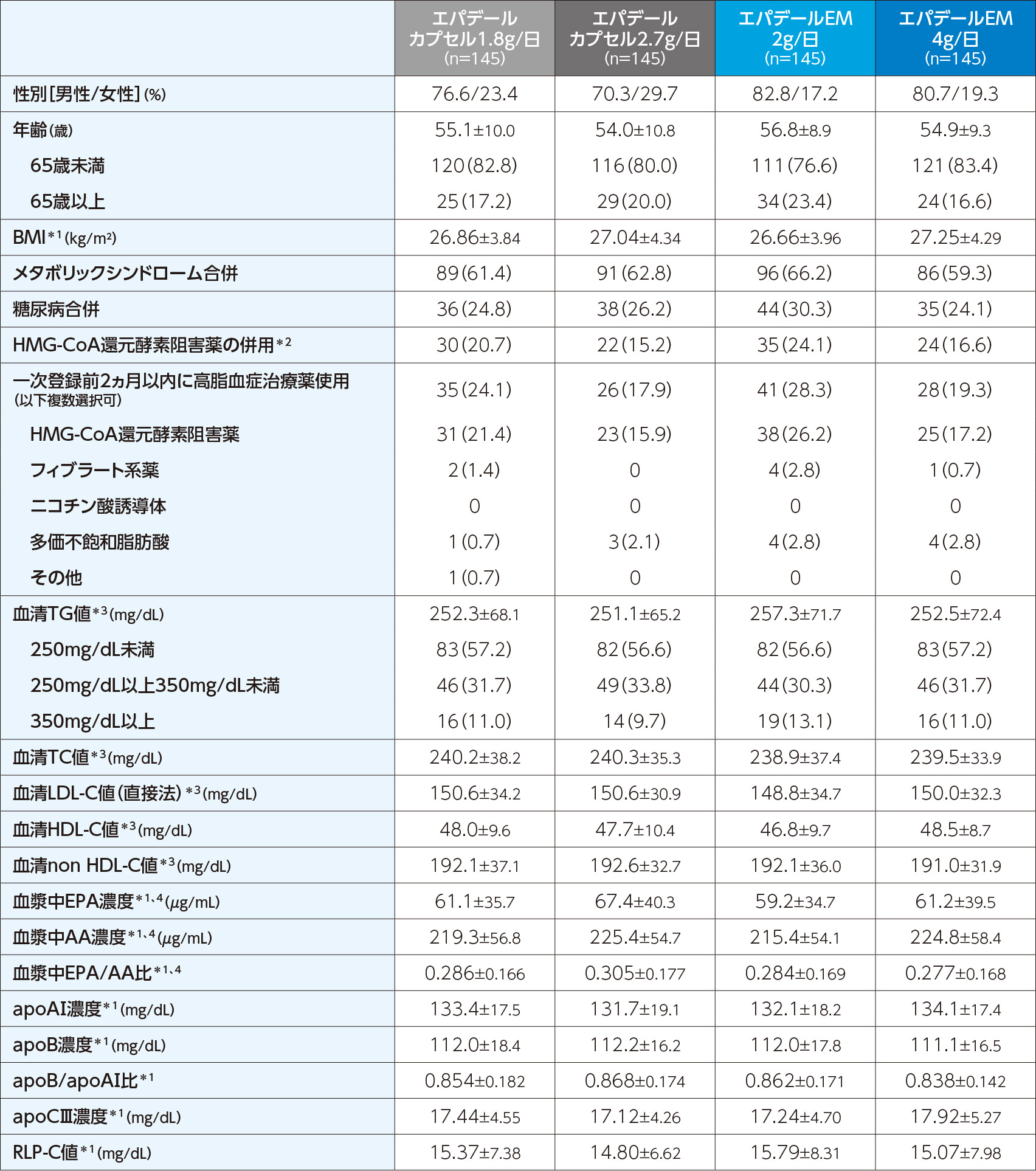
対象
生活習慣の改善指導を受けている高脂血症(TG高値)患者580例
- <選択基準>
-
- 同意取得時の年齢:20歳以上~75歳未満
- 前観察期の空腹時血清TG値150mg/dL以上500mg/dL未満(投与開始前8週時、4週時、2週時)
- 前観察期において血清TG値が安定している患者(「投与開始前8週時と4週時の空腹時血清TG値の差が、高い方の値の40%以内」かつ「投与開始前4週時と2週時の空腹時血清TG値の差が、高い方の値の30%以内」)
本試験では前観察期および投与期において、スタンダードスタチン(シンバスタチン、プラバスタチンナトリウム、フルバスタチンナトリウム)、β1遮断薬、β1選択的遮断薬、選択的β1アンタゴニスト、糖尿病治療薬の併用は、1日処方量の変更、薬剤の追加および変更がない限り可としたが、ストロングスタチン(アトルバスタチンカルシウム水和物、ピタバスタチンカルシウム水和物、ロスバスタチンカルシウム)を含む高脂血症治療薬、インスリン、エストロゲン、副腎皮質ホルモン(全身投与用)、レチノイド(外用剤を除く)、サイアザイド、上記以外のβ遮断薬、インターフェロンの併用は不可とした。
試験デザイン
第Ⅲ相、多施設共同、ランダム化、二重盲検、実薬対照、並行群間比較試験(ダブルダミー法)
投与方法
前観察期の評価で適格と判断された患者を、エパデールEM 2g/日群、4g/日群、エパデールカプセル1.8g/日群、2.7g/日群に1:1:1:1の割合で割り付け、エパデールEM 2g/日群、4g/日群では、それぞれエパデールEM 2g、4gを1日1回(朝)、エパデールカプセル1.8g/日群ではエパデールカプセル900mgを1日2回(朝・夕)、エパデールカプセル2.7g/日群ではエパデールカプセル900mgを1日3回(朝・昼・夕)、食直後に12週間経口投与した。
評価項目
◎主要評価項目
- <有効性>
-
投与12週後※1における投与前値※2からの血清TG値の変化率(検証的解析項目)
- <安全性>
-
投与開始時以降の有害事象
◎副次評価項目
- <有効性>
-
以下の項目に関する投与12週後※1における投与前値※2からの変化率
- ・血清総コレステロール(TC)値
- ・血清低比重リポ蛋白コレステロール(LDL-C)値(直接法)
- ・血清高比重リポ蛋白コレステロール(HDL-C)値
- ・血清non HDL-C値
- <安全性>
- 投与開始時以降の副作用
◎その他の評価項目
投与開始時および投与12週後または投与終了時(LOCF;Last Observation Carried Forward)における血漿中イコサペント酸(EPA)濃度、アラキドン酸(AA)濃度、EPA/AA比 など
解析計画
有効性の主要な解析対象集団は最大の解析対象集団(FAS)、安全性解析対象集団は治験薬が1回以上投与され、安全性の評価に関するデータが1つ以上ある患者とした。
◎主要評価項目
- <有効性>
-
投与12週後※1における投与前値※2からの血清TG値の変化率について共分散分析を行い、エパデールEM 2g/日のエパデールカプセル1.8 g/日に対する非劣性、ならびにエパデールEM 4g/日のエパデールEM 2g/日に対する優越性の2つの仮説について、閉検定手順を用いてそれぞれ検証した。閉検定手順はエパデールEM 2g/日のエパデールカプセル1.8g/日に対する非劣性が検証された場合に限り、エパデールEM 4g/日のエパデールEM 2g/日に対する優越性を検証することとした。
- ・エパデールEM 2g/日のエパデールカプセル1.8g/日に対する非劣性
-
投与群を説明変数、投与前値※2を共変量とした共分散分析を行い、投与群間差の95%信頼区間を求めた。エパデールEM 2g/日群とエパデールカプセル1.8g/日群の投与群間差の95%信頼区間の上限値が7%を下回った場合に、エパデールEM 2g/日はエパデールカプセル1.8g/日に対して非劣性であると判断することとした。
- ・エパデールEM 4g/日のエパデールEM 2g/日に対する優越性
-
投与群を説明変数、投与前値※2を共変量とした共分散分析を行い、エパデールEM 4g/日群とエパデールEM 2g/日群を比較した。
- <安全性>
- 投与群ごとに、投与開始時から後観察期の調査終了までに発現した有害事象の発現率を算出した。なお、エパデールEMの有効成分〔イコサペント酸エチル(EPA-E)〕は、抗血小板作用を有することから出血関連有害事象も集計した。
◎副次評価項目
- <有効性>
- 投与群を説明変数、投与前値※2を共変量とした共分散分析を行い、エパデールEM 2g/日群とエパデールカプセル1.8g/日群、エパデールEM 4g/日群とエパデールEM 2g/日群を比較した。なお、血清non HDL-C値についてはエパデールEM 4g/日群とエパデールEM 2g/日群の有意差検定も実施した。
- <安全性>
- 投与群ごとに、投与開始時から後観察期の調査終了までに発現した副作用の発現率を算出した。有害事象と同様に出血関連副作用も集計した。
◎その他の評価項目
投与終了時の結果はLOCFで補完した。
- ※1 投与10週後および12週後の測定値の平均値(投与中止例の場合は評価可能な最終2時点の測定値の平均値を用い、最終2時点の間隔が20日間を超えている場合は、評価可能な最終時点の測定値を用いた。治験薬投与後に1時点の測定値しかない場合は、その時点の測定値を用いた。)
- ※2 投与開始前4週時、2週時および投与開始時の測定値の平均値