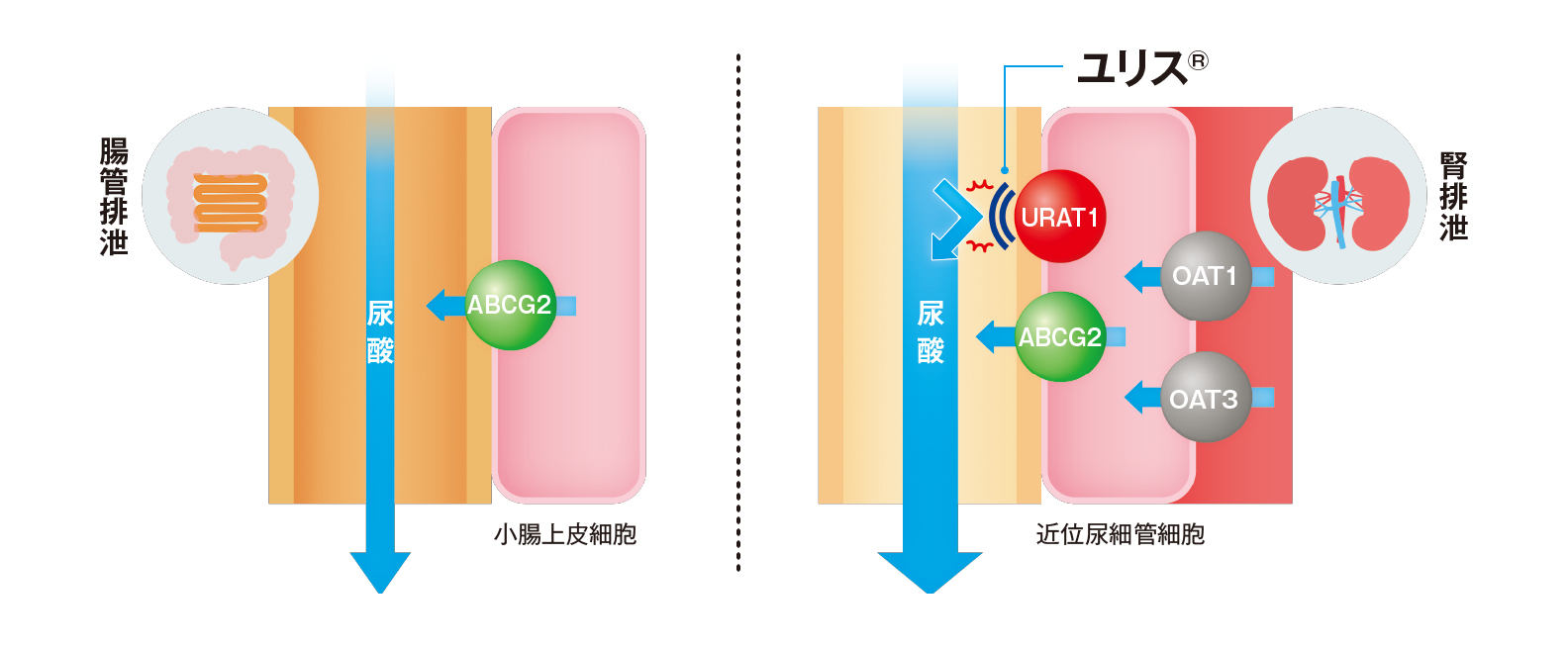
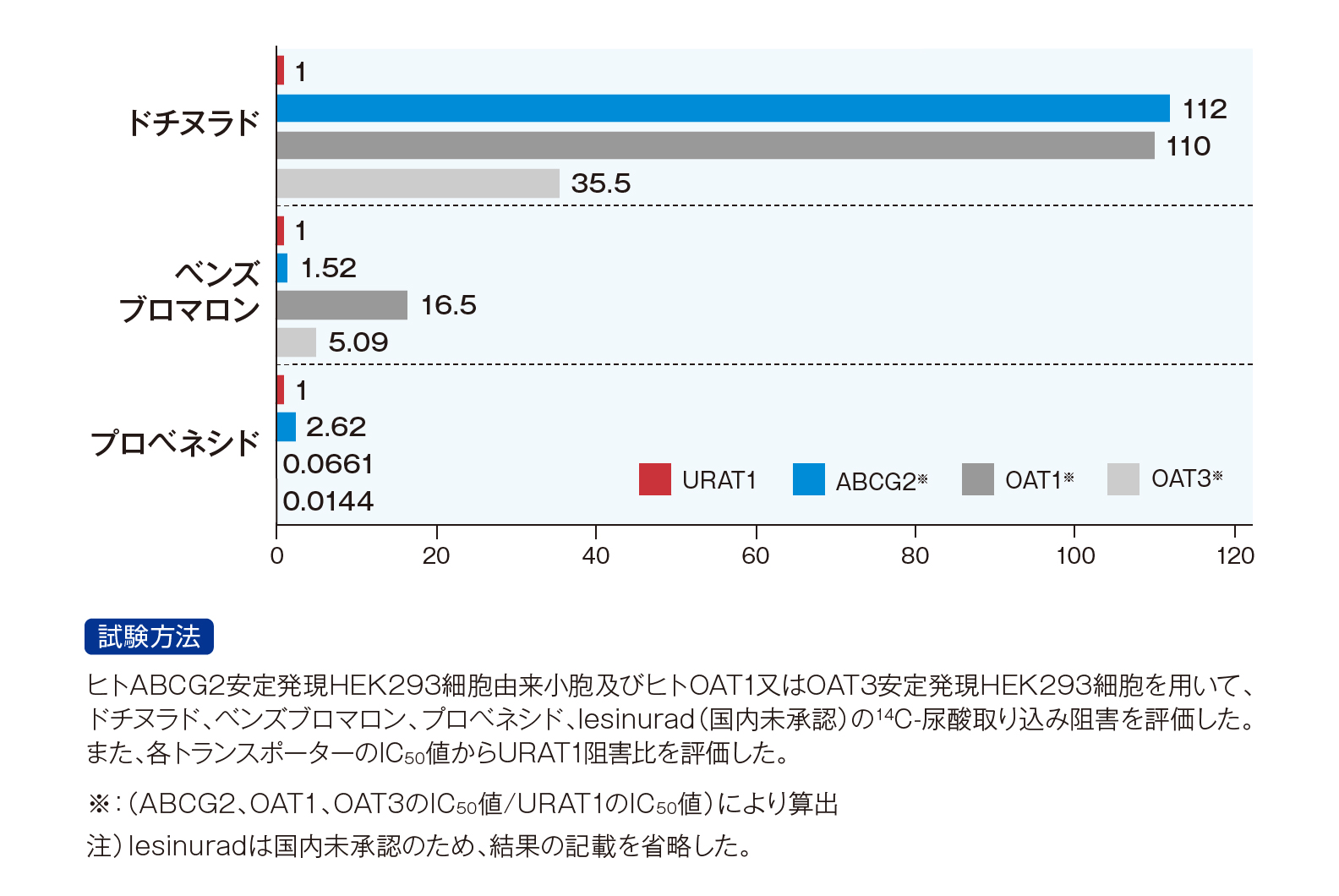
「禁忌を含む注意事項等情報」等は製品情報(DI)をご参照ください。
ユリス®錠の血清尿酸値低下作用
[後期第Ⅱ相試験(用量反応検証試験)]
後期第Ⅱ相試験は一部承認外の用法及び用量による成績が含まれますが、用量反応検証試験として実施されたため掲載します。
社内資料:用量反応検証試験・後期第Ⅱ相臨床試験[2020年1月23日承認、CTD2.7.6.14、CSR FYU-981-006(資料5.3.5.1-2)]〔承認時評価資料〕
Hosoya T, et al. Clin Exp Nephrol 2020; 24: S53-61
[利益相反]本研究は株式会社富士薬品の資金により行われた。本論文の著者のうち4名は株式会社富士薬品の社員である。著者には、本研究に関する株式会社富士薬品のアドバイザーでありコンサルタント料等を受領している者が含まれる。
後期第Ⅱ相試験(用量反応検証試験)の試験概要
-
【目 的】
痛風を含む高尿酸血症患者を対象として、ユリスの用量反応性の検証及び安全性の検討を行う。
-
【試験デザイン】
多施設共同、二重盲検、ランダム化、プラセボ対照、用量漸増並行群間比較試験
-
【対 象】
20歳以上の痛風を含む高尿酸血症患者※1(割付症例数:201、投与症例数:200)[血清尿酸値:痛風患者7.0mg/dL以上、高尿酸血症患者(合併症※2あり)8.0mg/dL以上、高尿酸血症患者(合併症※2なし)9.0mg/dL以上]
※1 「尿酸産生過剰型」あるいは「判定不能」を除く
※2 高血圧、糖尿病、メタボリックシンドローム
-
【方 法】
対象患者をユリス4用量群(0.5mg群、1mg群、2mg群、4mg群)又はプラセボ群の計5群にランダムに割り付け、治験薬を1日1回朝食後に経口投与した。ユリス4用量群は0.25mg/日を投与開始から2週後まで2週間投与した後、0.5mg/日に増量して3週目から4週後まで2週間投与し、さらに0.5mg/日、1mg/日、2mg/日、4mg/日のいずれかに維持又は増量して5週目から12週後まで8週間投与した。
-
【評 価 項 目】
有効性に関する評価項目:
[主要評価項目]投与終了時(投与12週後又は投与中止時)における投与前値からの血清尿酸値低下率[(投与前値-投与終了時の値)/投与前値×100]
[副次評価項目]投与終了時における投与前値からの血清尿酸値変化量、投与終了時の⾎清尿酸値6.0mg/dL以下の達成率など
安全性に関する評価項目:有害事象、副作用、痛風関節炎など
-
【解 析 計 画】
主要評価項目について、投与終了時の血清尿酸値低下率は、Jonckheere-Terpstra検定を用いた用量反応性の検証(主解析)及びTukey-Kramer検定を用いた群間比較を行った。
副次評価項目について、血清尿酸値変化量(投与終了時)は、Jonckheere-Terpstra検定を用いた用量反応性の検討及びTukey-Kramer検定を用いた群間比較を行った。血清尿酸値6.0mg/dL以下の達成率は、投与終了時におけるCochran-Armitage検定を用いた用量反応性の検討を行った。
投与終了時の血清尿酸値低下率(主要評価項目;FAS解析対象、LOCF)
ユリス0.5mg群~4mg群において、
投与終了時の血清尿酸値低下率に用量反応性が検証されました。
投与終了時における投与前値からの血清尿酸値低下率(平均値±標準偏差)は、プラセボ群で-2.83±8.19%、ユリス0.5mg群で21.81±11.35%、1mg群で33.77±9.82%、2mg群で42.66±13.16%、4mg群で61.09±8.75%であり、ユリスの用量反応性が検証されました([主解析]p<0.001、Jonckheere-Terpstra検定)。また、群間比較では、いずれの群間においても有意差が認められました(ユリス1mg群vs.2mg群:名目上のp=0.002、その他の群間:名目上のp<0.001、Tukey-Kramer検定)。
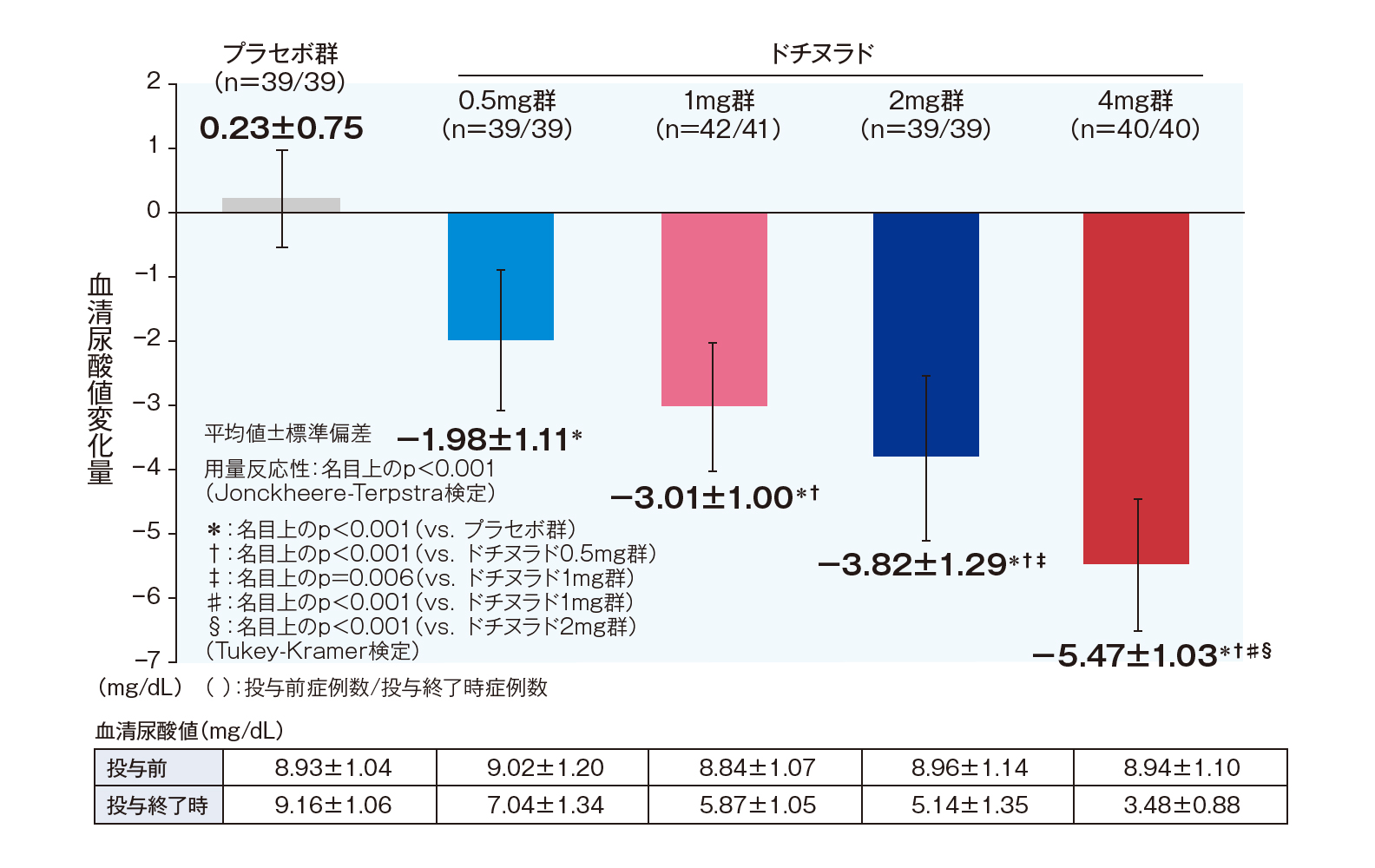
投与終了時の投与前値からの血清尿酸値変化量(副次評価項目;FAS解析対象、LOCF)
投与終了時における投与前値からの血清尿酸値変化量(平均値±標準偏差)は、プラセボ群で0.23±0.75mg/dL、ユリス0.5mg群で-1.98±1.11mg/dL、1mg群で-3.01±1.00mg/dL、2mg群で-3.82±1.29mg/dL、4mg群で-5.47±1.03mg/dLであり、ユリスの用量反応性が認められました(名目上のp<0.001、Jonckheere-Terpstra検定)。
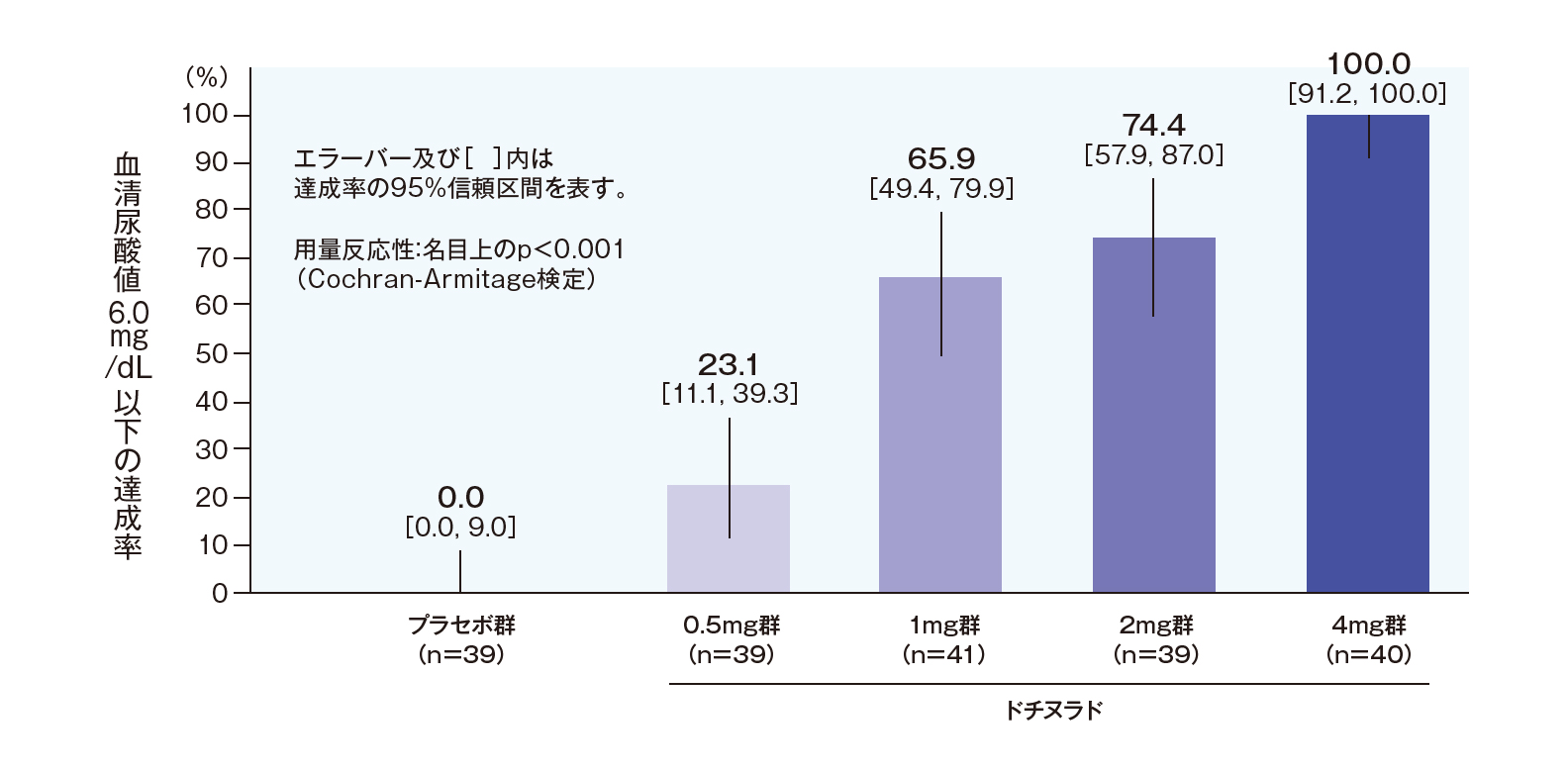
投与終了時の血清尿酸値6.0mg/dL以下の達成率(副次評価項目;FAS解析対象、LOCF)
投与終了時の血清尿酸値6.0mg/dL以下の達成率は、プラセボ群で0.0%、ユリス0.5mg群で23.1%、1mg群で65.9%、2mg群で74.4%、4mg群で100.0%であり、ユリスの用量反応性が認められました(名目上のp<0.001、Cochran-Armitage検定)。
安全性(SP解析対象)
副作用は、プラセボ群で15.4%、
ユリス0.5mg群で15.0%、1mg群で19.0%、2mg群で17.9%、4mg群で17.5%に認められました。
副作用は、プラセボ群で6/39例(15.4%)、ユリス0.5mg群で6/40例(15.0%)、1mg群で8/42例(19.0%)、2mg群で7/39例(17.9%)、4mg群で7/40例(17.5%)に認められました。2例以上に認められた副作用は、プラセボ群で尿中β2ミクログロブリン増加が2/39例(5.1%)、ユリス1mg群でβ-NアセチルDグルコサミニダーゼ増加が3/42例(7.1%)、痛風関節炎が2/42例(4.8%)、2mg群で痛風関節炎が3/39例(7.7%)、尿中β2ミクログロブリン増加、β-NアセチルDグルコサミニダーゼ増加、白血球数増加が各2/39例(5.1%)、4mg群で痛風関節炎が3/40例(7.5%)、尿中β2ミクログロブリン増加、β-NアセチルDグルコサミニダーゼ増加、α1ミクログロブリン増加が各2/40例(5.0%)に認められました。ユリス0.5mg群で認められた副作用は、痛風関節炎、四肢不快感、γ-グルタミルトランスフェラーゼ増加等でいずれも1/40例(2.5%)に認められました。本試験において、死亡例を含む重篤な副作用は認められませんでした。また本試験において投与中止に至った副作用は、プラセボ群で尿中β2ミクログロブリン増加が、1mg群で痛風関節炎がそれぞれ1例に認められました。
【FAS解析対象】:治験薬を1回以上投与され、有効性に関する評価項目が投与後に1項目でも測定された症例
【LOCF】:欠測データを最後に観察した値に置き換えて補完する
【SP解析対象】:治験薬を1回以上投与され、投与後に安全性の評価が可能な情報が得られている症例
6. 用法及び用量
通常、成人にはユリスとして1日0.5mgより開始し、1日1回経口投与する。その後は血中尿酸値を確認しながら必要に応じて徐々に増量する。維持量は通常1日1回2mgで、患者の状態に応じて適宜増減するが、最大投与量は1日1回4mgとする。
▲TOPへ戻る
ユリス®錠の長期投与における血清尿酸値低下作用
[第Ⅲ相試験(長期投与試験)]
社内資料:第Ⅲ相長期投与試験[2020年1月23日承認、CTD 2.7.6.17、CSR FYU-981-010(資料5.3.5.2-1)]〔承認時評価資料〕
Hosoya T, et al. Clin Exp Nephrol 2020; 24: S80-91
[利益相反]本研究は株式会社富士薬品の資金により行われた。本論文の著者のうち4名は株式会社富士薬品の社員である。著者には、本研究に関する株式会社富士薬品のアドバイザーでありコンサルタント料等を受領している者が含まれる。
第Ⅲ相試験(長期投与試験)の試験概要
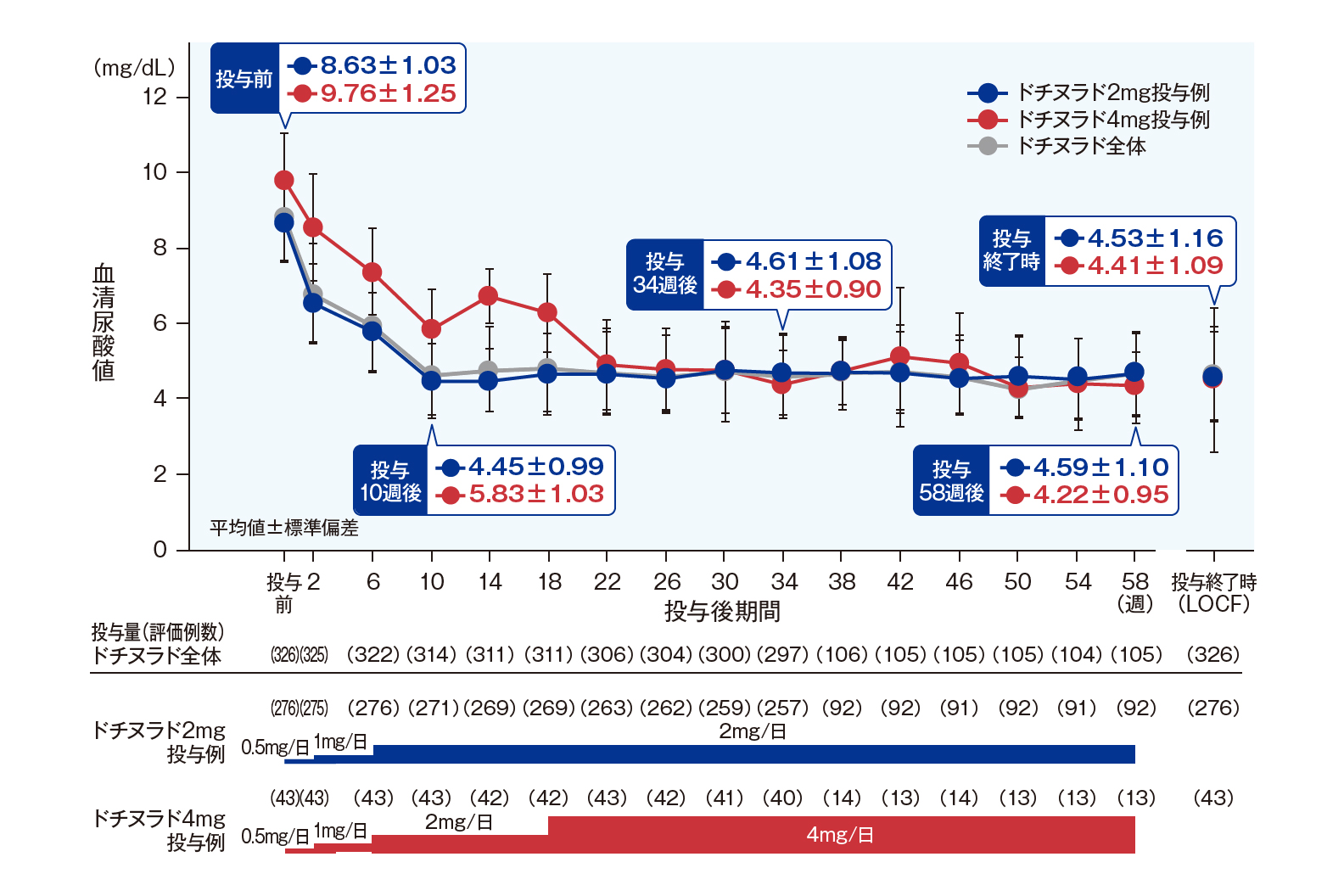
各時点の血清尿酸値低下率(主要評価項目;FAS解析対象)
投与10週後から投与58週後まで、
ユリス全体の血清尿酸値低下率の平均値は44.99%~49.71%の間で推移しました。
各時点(投与2週後以降、58週後まで4週ごと)における投与前値からの血清尿酸値低下率(平均値±標準偏差)は、投与34週後では、ユリス全体で47.83±10.85%、2mg投与例で46.73±10.77%、4mg投与例で54.92±8.58%、投与58週後では、ユリス全体で48.43±11.38%、2mg投与例で47.17±11.18%、4mg投与例で57.35±8.73%でした。投与10週後から投与58週後まで、ユリス全体の血清尿酸値低下率の平均値は44.99%~49.71%の間で推移しました。いずれの投与量も、全ての時点で、投与前値との比較において有意差が認められました(名目上のp<0.001、1標本t検定)。
各時点及び投与終了時の血清尿酸値(副次評価項目;FAS解析対象)
投与58週後では、ユリス全体で4.55±1.09mg/dL、
2mg投与例で4.59±1.10mg/dL、4mg投与例で4.22±0.95mg/dLでした。
各時点(投与2週後以降、58週後まで4週ごと)及び投与終了時における血清尿酸値(平均値±標準偏差)は、下図のとおりに推移し、投与34週後では、ユリス全体で4.57±1.06mg/dL、2mg投与例で4.61±1.08mg/dL、4mg投与例で4.35±0.90mg/dL、投与58週後では、ユリス全体で4.55±1.09mg/dL、2mg投与例で4.59±1.10mg/dL、4mg投与例で4.22±0.95mg/dLでした。
安全性(SP解析対象)
副作用は、ユリス2mg投与例で18.8%、4mg投与例で34.9%に認められました。
副作用は、ユリス2mg投与例で52/277例(18.8%)、4mg投与例で15/43例(34.9%)に認められました。2例以上に認められた副作用は、ユリス2mg投与例で痛風関節炎が30/277例(10.8%)、関節炎が7/277例(2.5%)、四肢不快感が3/277例(1.1%)、腎石灰沈着症、腎結石症、血中クレアチニン増加が各2/277例(0.7%)、4mg投与例※で痛風関節炎が9/43例(20.9%)、四肢不快感が4/43例(9.3%)、腎結石症が3/43例(7.0%)、尿中アルブミン/クレアチニン比増加が2/43例(4.7%)に認められました。
※:投与19週目に4mgへ増量
本試験において、死亡例は認められませんでした。本試験において、重篤な副作用はユリス2mg投与時に胃癌第1期が1例に認められました。本試験において投与中止に至った副作用は、ユリス0.5mg投与時にそう痒症、軟便、尿中β2ミクログロブリン増加、アラニンアミノトランスフェラーゼ増加、γ-グルタミルトランスフェラーゼ増加が各1例、1mg投与時に痛風関節炎、アスパラギン酸アミノトランスフェラーゼ増加が各1例、2mg投与時に腎結石症が2例及び湿疹が1例、4mg投与時に腎結石症が3例に認められました。
【FAS解析対象】:治験薬を1回以上投与され、有効性に関する評価項目が投与後に1項目でも測定された症例
【LOCF】:欠測データを最後に観察した値に置き換えて補完する
【SP解析対象】:治験薬を1回以上投与され、投与後に安全性の評価が可能な情報が得られている症例
▲TOPへ戻る
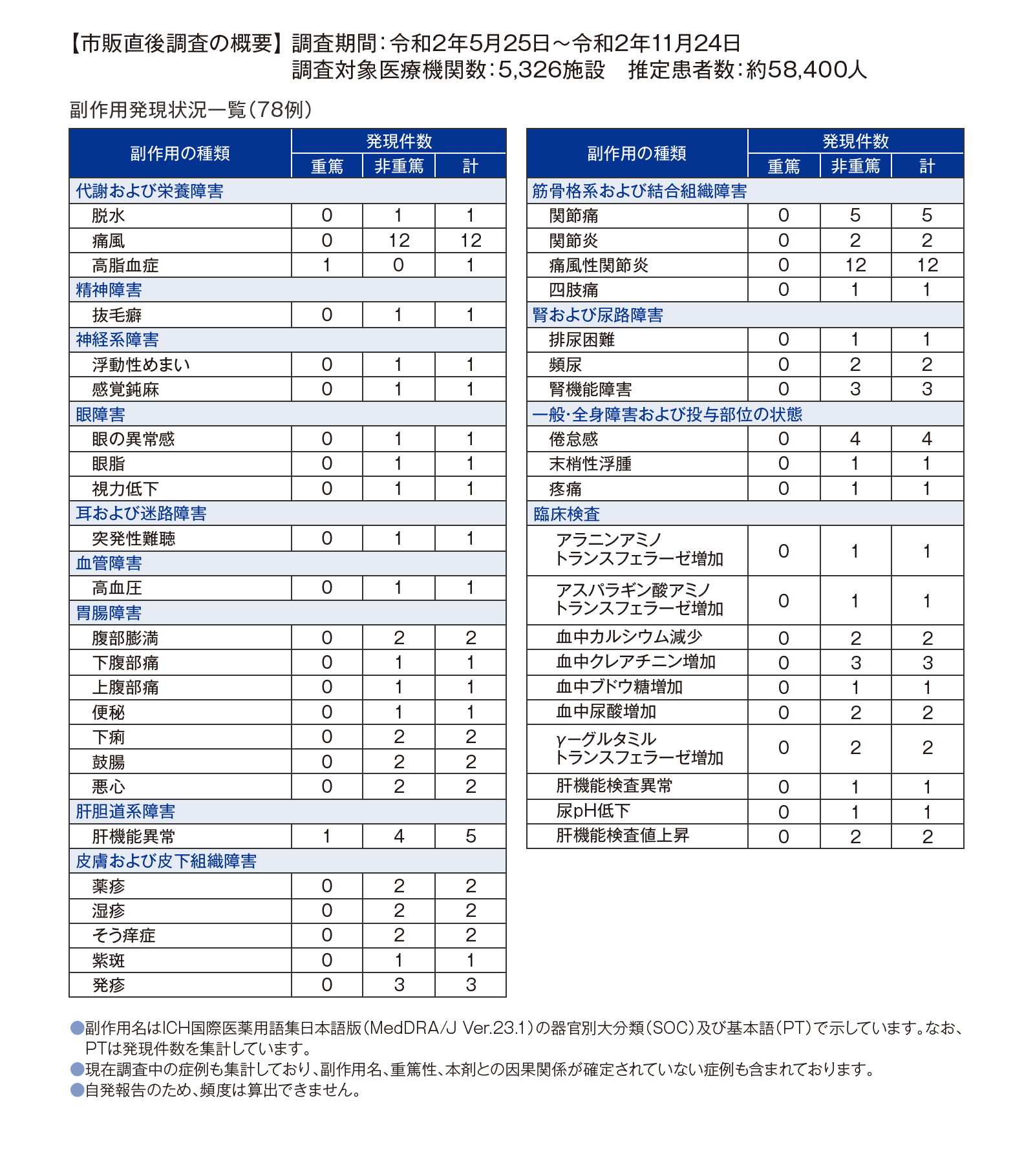
発売後6ヵ月間の副作用発現状況
[ユリス®錠 市販直後調査]
ユリス®錠 市販直後調査期間中の副作用最終集計結果のご報告
副作用最終集計結果
調査対象5,326施設において、推定患者数約58,400人にユリスが投与され、78例96件の副作用が収集されました。
そのうち重篤な副作用は、「高脂血症」「肝機能異常」各1件(同一症例で発現)でした。
 ▲TOPへ戻る
▲TOPへ戻る
GFR区分別の高尿酸血症患者における
ユリスの有効性および安全性の検討
(GFR区分別の検討):後ろ向き観察研究
嶋津啓二, 他 診療と新薬 2024 ; 61 : 689-700
[利益相反]本研究は持田製薬株式会社及び株式会社富士薬品の資金により行われた。
試験概要
-
【目 的】
高尿酸血症患者〔腎機能が糸球体濾過量(GFR)区分としてG1~G5〕における、ユリスの使用実態と尿酸降下作用および腎機能に与える影響を評価する。
-
【試験デザイン】
-
【対 象】
調査対象期間(情報抽出対象期間:2019年1月1日~2023年11月30日)中に大阪府済生会中津病院(以下、当院)においてユリスを投与された患者のうち、以下の適格基準を満たし、かつ、添付文書の用法及び用量に従って投与された症例のうち1つ以上の評価項目データを有し、投与開始時の血清尿酸値が6.0mg/dLより大きい患者298例。
【適格基準】ユリス投与開始時点でGFR区分G1~G5に分類可能、かつ、20歳以上の患者(性別不問)※。
※:他の医療機関でユリスを投与開始された後に当院を受診した患者(当院の初回受診時にユリスを既に内服している患者)、オプトアウトにより不参加の意思表示のあった患者は除外した。
-
【方 法】
調査対象期間(2019年1月1日~2023年11月30日)の診療録から適格基準を満たす患者を検索し、ユリスの初回投与日を起点日として、前後18ヵ月の範囲内のデータを解析した。
-
【評 価 項 目】
[主要評価項目]投与開始後12ヵ月時の血清尿酸値6.0mg/dL以下達成割合(GFR区分別のサブグループ解析も実施)
[その他の評価項目]※※投与開始後3、6、12ヵ月における血清尿酸値※1、投与開始前後各12ヵ月間の推算糸球体濾過量(eGFR)スロープの変化※2 等
※※:その他の評価項目は、解析可能症例にて実施。
※1:投与開始時ステータス別※3のサブグループ解析も実施。
※2:GFR区分別および投与開始時ステータス別※3のサブグループ解析も実施。
※3:投与開始前3ヵ月間に他の尿酸降下薬の投与が一度もない症例を「新規」、投与開始前3ヵ月間に他の尿酸降下薬の投与が1回以上ありユリス投与開始時に同時投与のない症例を「切替」、投与開始前3ヵ月間に他の尿酸降下薬の投与が1回以上ありユリス投与開始時に1剤以上の同時投与のある症例を「併用」と定義。
[安全性評価項目]イベント(痛風発作、心血管合併症、透析導入)の発生/肝機能(AST、ALT、γ-GTP)の変化
-
【解 析 計 画】
12ヵ月時点における血清尿酸値6.0mg/dL以下達成割合(主要評価項目)についての解析は、各時点の血清尿酸値6.0mg/dL以下達成の有無を従属変数とし、ベースライン血清尿酸値を共変量とし、投与開始後の日数を表す変数(連続値)を非線形の説明変数として加えた一般化推定方程式を実施した。また、GFR区分による傾向の違いを検討するために、主要評価項目についての解析に、投与開始時点におけるGFR区分(G1、2、G3a、G3b、G4、G5)と時点を表す変数との交互作用項を含むモデルを用いたサブグループ解析を実施した。
投与開始後の血清尿酸値の推移については、各時点(3、6、12ヵ月)の値を従属変数とし、ベースライン値を共変量とし、時間(3、6、12ヵ月)を表す変数(カテゴリー変数)を説明変数として加えた線形混合効果モデルを実施した。また、サブグループ解析では、開始時ステータスによる傾向の違いを検討するために、前述の解析に、投与開始時点における開始時ステータスと時点を表す変数との交互作用項を含むモデルを用いた解析を実施した。
投与開始前後各12ヵ月間のeGFRのスロープについては、ユリス投与開始日を起点に、前後12ヵ月間のeGFRを従属変数、時間とユリス投与との交互作用項を含む線形混合効果モデルを用いて、前後のスロープの違いを検討した。また、GFR区分、投与開始時ステータスで分けたサブグループ解析も実施した。サブグループ解析ではグループ間の比較はしなかった。
研究の限界
後ろ向き観察研究である。ユリス非投与群を同時対照とした無作為化比較試験ではないため、得られた情報に制限があった。とはいえ、診療録からの情報を電子的に抽出して用いることによって、検査データをすべて利用することができただけでなく、脱落や欠測等の影響を最小限にすることが可能であった。更には、ユリス投与開始前後のデータを利用できたことから、背景調整のための情報が不十分であるという問題を、同一症例内の前後比較によって解決することが可能であった。
正確なユリス投与量が得られなかった。本研究対象におけるユリス投与開始日は、長期投与が許可される前の期間を含み、投与量と指示された内服量が実際には異なる症例が含まれることが想定される。また、アドヒアランスについての情報も得ることができなかったため、投与量とアウトカムとの関係は検討することができなかった。
単施設のみの結果である。本研究結果の一般化に限界があることは否定できないが、ユリス投与実績においては現解析時点における本邦で最大規模のデータであり、解析結果も十分な検出力のもとで得られたと考えられる。
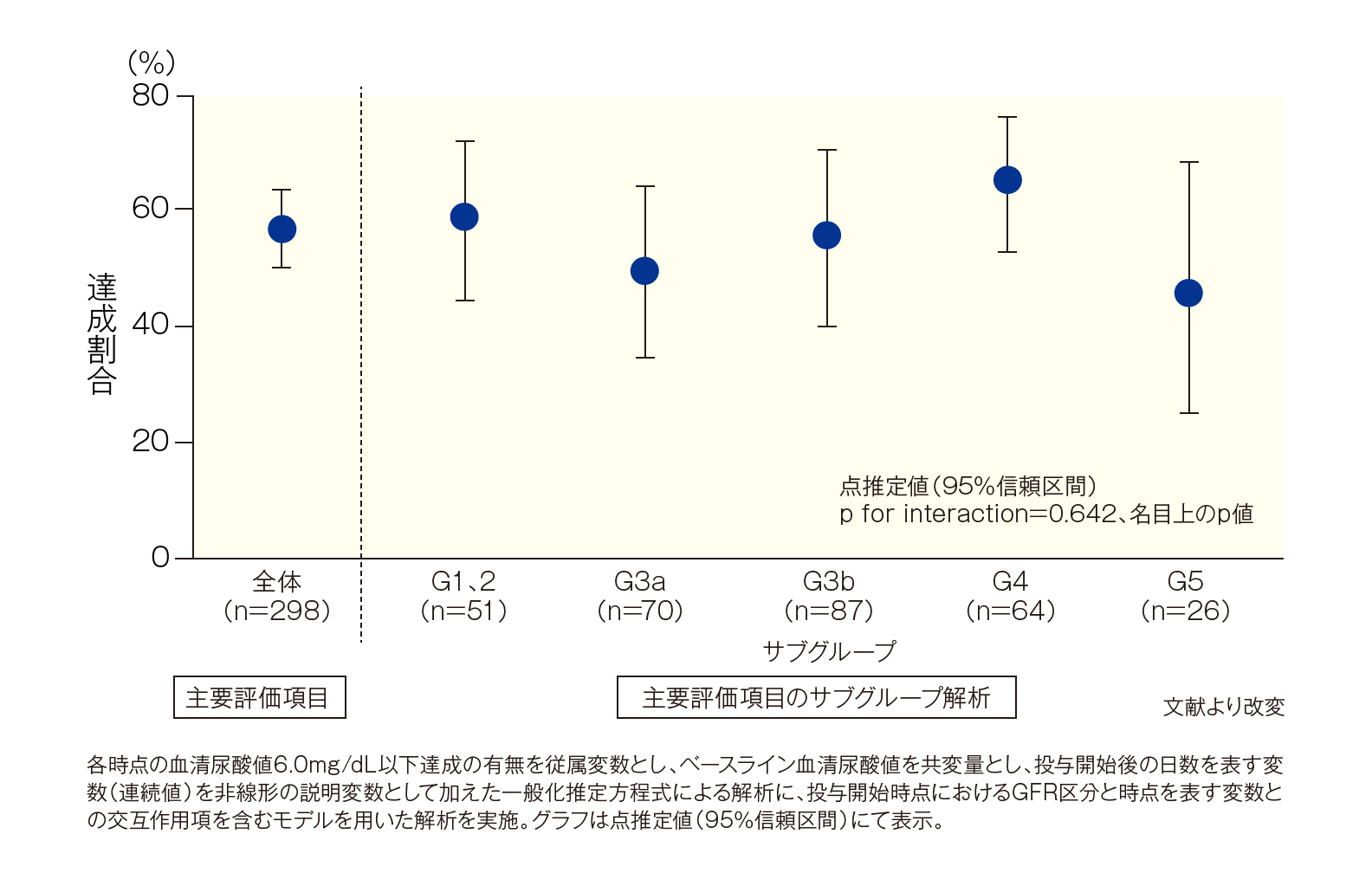
[主要評価項目]血清尿酸値6.0mg/dL以下達成割合(ユリス投与12ヵ月後)
全体(主要評価項目)では、投与開始後12ヵ月時点における血清尿酸値6.0mg/dL以下達成割合の点推定値(95%信頼区間)は56.8(50.0-63.4)%でした。
投与開始時のGFR区分別(サブグループ解析)では、G1、2で58.7(44.4-71.7)%、G3aで49.4(34.8-64.1)%、G3bで55.8(40.0-70.4)%、G4で65.3(52.8-75.9)%、G5で46.0(25.1-68.4)%でした。投与開始時のGFR区分による傾向の違いは認めませんでした[p for interaction=0.642(名目上のp値)、一般化推定方程式による解析に、投与開始時点におけるGFR区分と時点を表す変数との交互作用項を含むモデルを用いた解析を実施]。
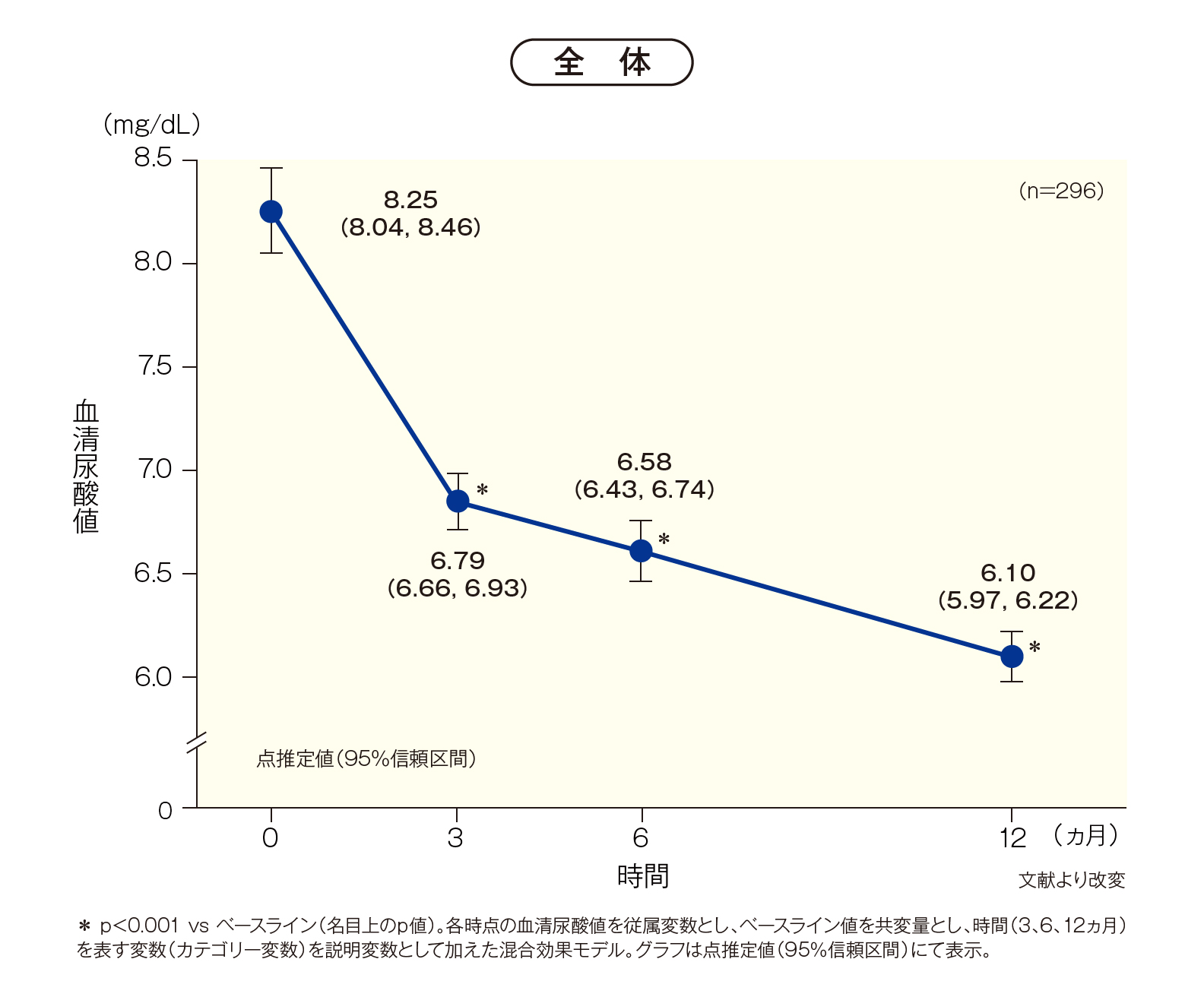
[その他の評価項目]血清尿酸値の推移
全体の血清尿酸値の点推定値(95%信頼区間)は、投与開始時8.25(8.04-8.46)mg/dLから、
3ヵ月後6.79(6.66-6.93)mg/dL、6ヵ月後6.58(6.43-6.74)mg/dL、12ヵ月後6.10(5.97-6.22)mg/dLへと
有意に低下しました[p<0.001 vsベースライン(名目上のp値)、線形混合効果モデル]。
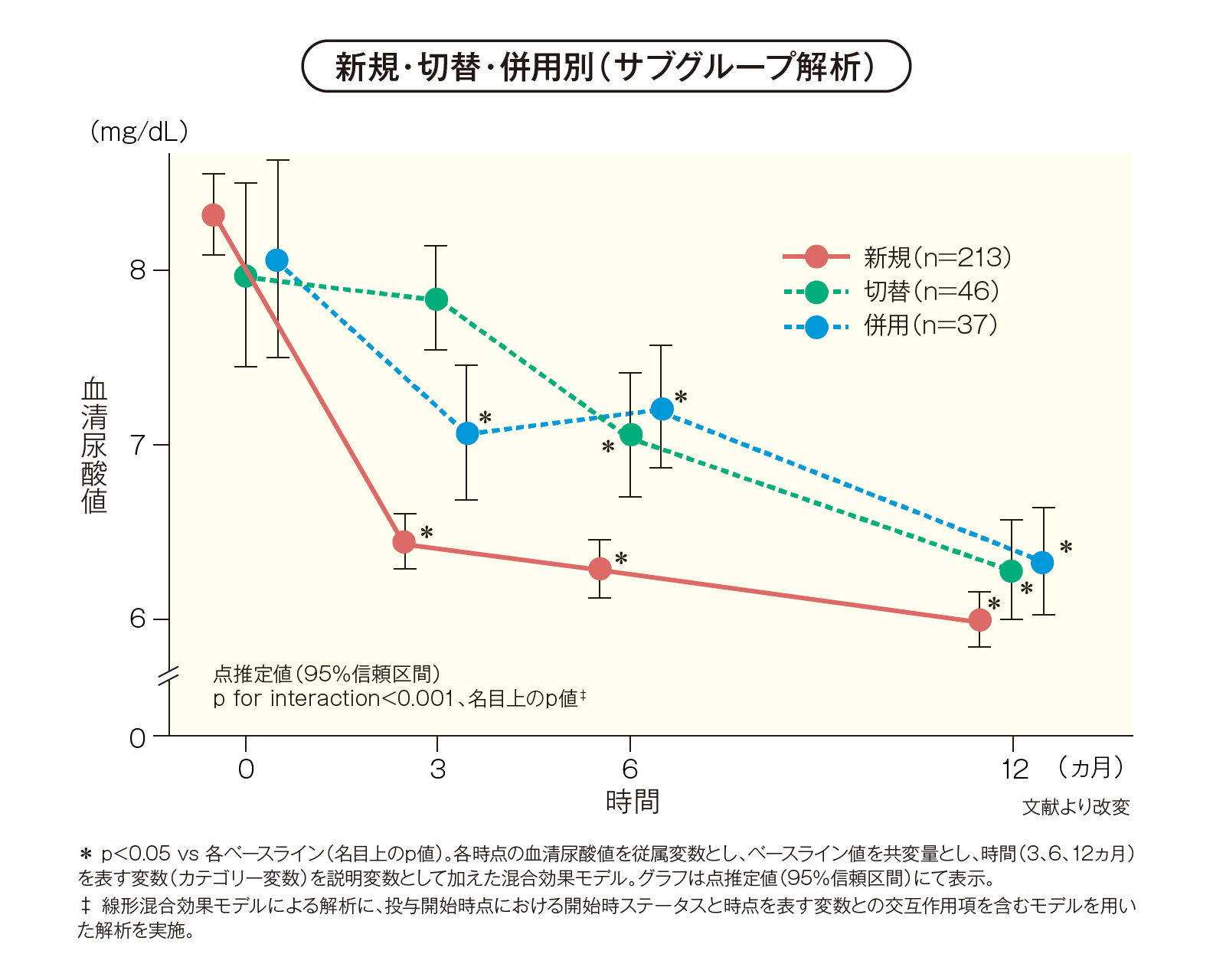
投与開始時ステータス別では、新規症例は投与開始時8.34(8.09-8.58)mg/dLから12ヵ月後5.98(5.83-6.14)mg/dLへ、切替症例は投与開始時7.98(7.45-8.51)mg/dLから12ヵ月後6.27(5.98-6.56)mg/dLへ、併用症例は投与開始時8.07(7.49-8.64)mg/dLから12ヵ月後6.32(6.01-6.63)mg/dLへと有意に低下しました[p<0.05 vs 各ベースライン(名目上のp値)、線形混合効果モデル]。
また、新規、切替、併用による傾向の違いが認められました[p for interaction<0.001(名目上のp値)、線形混合効果モデルによる解析に、投与開始時点における開始時ステータスと時点を表す変数との交互作用項を含むモデルを用いた解析を実施]。
[安全性評価項目]イベントの発生/肝機能の変化
痛風発作は1.7%、心血管合併症は2.0%、透析導入は6.0%でした。
いずれもユリスとの因果関係はありませんでした。
投与開始後の肝機能の変化は、下表のとおりでした。
イベント(痛風発作、心血管合併症、透析導入)の発生
痛風発作は5例(1.7%)、心血管合併症は6例(2.0%)、透析導入は18例(6.0%)であった。いずれもユリスとの因果関係はなかった。
肝機能(AST、ALT、γ-GTP)の変化
投与開始後の肝機能の変化は下記のとおりであった。
点推定値(95%信頼区間)
| 肝機能パラメータ |
開始時 |
12ヵ月後 |
| AST(U/L) |
26.1(21.7-30.5) |
26.6(24.2-29.0) |
| ALT(U/L) |
21.5(18.2-24.8) |
21.1(19.3-22.9) |
| γ-GTP(U/L) |
48.8(39.1-58.6) |
49.9(44.5-55.3) |
文献より作表
本研究で収集した安全性情報は上記項目のみであり、それ以外の安全性情報に関するデータは評価されませんでした。ユリスの注意事項等情報や安全性情報につきましては、製品情報(DI)をご参照ください。
9. 特定の背景を有する患者に関する注意(一部抜粋)
9.2 腎機能障害患者 9.2.1 重度の腎機能障害患者(eGFRが30mL/min/1.73m2未満)
他剤での治療を考慮すること。本剤は腎近位尿細管において作用するため、腎機能障害の程度に応じて、有効性が減弱する可能性がある。特に、乏尿又は無尿の患者においては、有効性が期待できないことから、本剤の投与は避けること。
なお、臨床試験では、eGFRが30mL/min/1.73m2未満の患者は除外されている。
▲TOPへ戻る