- 医療関係者向けホーム
- 循環器領域
- トレプロスト®注射液
- Clinical Study:国内第Ⅱ/Ⅲ相試験(エポプロステノールからの切替え例を対象とした試験:皮下投与又は静脈内投与試験) 試験の概要
 Clinical Study国内第Ⅱ/Ⅲ相試験(エポプロステノールからの切替え例を対象
Clinical Study国内第Ⅱ/Ⅲ相試験(エポプロステノールからの切替え例を対象
とした試験:皮下投与又は静脈内投与試験)
承認外の用法及び用量が含まれますが、承認時評価資料のため紹介します。
試験の概要「国内第Ⅱ/Ⅲ相試験
(皮下投与又は静脈内投与試験;MD070123P21試験)1,2)
エポプロステノールからの切替え例を対象とした試験」
- 1)持田製薬社内資料:国内患者対象試験(2014年3月24日承認、CTD 2.7.6.7.1)<承認時評価資料>
- 2)大森庸子ほか:Prog Med 34, 333-348(2014)
[利益相反:共同執筆者(京谷晋吾)は、トレプロスチニルのPAHの治験の医学専門家を務めた。
また、著者に持田製薬株式会社の社員が含まれている。本論文の作成、出版に関する費用は持田製薬株式会社が負担した。]
「警告、禁忌を含む使用上の注意」等は電子添文をご参照ください。
試験デザイン
多施設共同、非盲検、非ランダム化、非対照試験
目的
エポプロステノールからトレプロスチニルへの切替え後における、トレプロスチニルの持続皮下投与及び持続静脈内投与の有効性、安全性及び薬物動態を検討した。
対象
16~64歳の特発性/家族性又は膠原病性血管疾患に伴うPAH患者18例
- WHO機能分類クラスⅡ~Ⅲ
- 6分間歩行試験:歩行距離≧250m
- 右心カテーテル検査:安静時平均肺動脈圧>25mmHg、肺毛細血管楔入圧<15mmHg、肺血管抵抗>3mmHg・分/L
投与方法
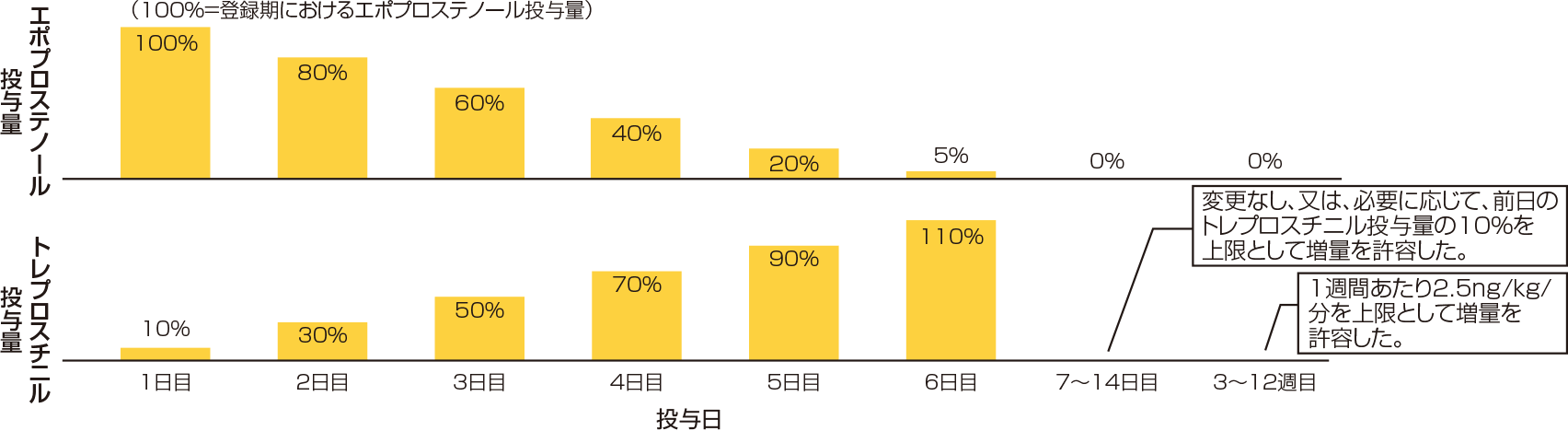
トレプロスチニルの用量を登録期のエポプロステノールの投与量の10%からとし、持続皮下投与又は持続静脈内投与した。有害事象の発現により皮下投与あるいは静脈内投与の継続が困難な場合のみ、投与経路の変更を許容した。その後、以下に示した投与量(推奨用量)調節スケジュールに従ってエポプロステノールの投与量を減少させながら、トレプロスチニルの投与量を増量し、原則2週間以内に切替えを完了した。その後は、臨床症状を考慮し、トレプロスチニルの投与量を増減した。増量は1週間当たり2.5ng/kg/分以下とした。減量する場合は、原則、1週間当たり2.5ng/kg/分以下とした。ただし、有害事象の発現やトレプロスチニルの投与を中止し、他のPAH治療の開始が必要となるなど、緊急を要する場合は、急激な減量に伴うリバウンドに注意しながら、1週間当たり2.5ng/kg/分を超えた減量を許容した。
また、持続皮下投与の場合は、腹部などに皮下投与し、静脈内投与の場合は、中心静脈カテーテルを留置し、中心静脈内へ投与した。ただし、緊急の場合のみ末梢静脈内への投与を許容した。
評価項目

略語一覧
- PVRI:肺血管抵抗係数
- CI:心係数
- PAPm:平均肺動脈圧
- HR:心拍数
- CO:心拍出量
- PAPs:収縮期肺動脈圧
- PAPd:拡張期肺動脈圧
- PCWP:肺毛細血管楔入圧
- RAPm:平均右心房圧
- SvO2:混合静脈血酸素飽和度
- SpO2:経皮的動脈血酸素飽和度
- SAPm:平均全身動脈圧
- PVR:肺血管抵抗
- SVR:体血管抵抗
- SVRI:体血管抵抗係数
- TPR:全肺抵抗
- TPRI:全肺抵抗係数
- SV:一回拍出量
- SI:一回拍出係数
安全性評価項目
有害事象、他
- a:10~12週の間で投与経路変更時の観察、検査及び評価を実施した場合、12週後の観察、検査及び評価の実施は不要。
- b:同意取得前6週間以内に6分間歩行試験を実施した経験がない場合は、6分間歩行の練習を実施してから行った。
- c:評価時期の設定のみで評価項目としての定義なし。
- d:臨床症状の悪化による次のいずれかのイベント:「併用禁止薬及び併用禁止療法の使用、又は併用禁止薬及び併用禁止療法使用のための中止」、「最初の入院」、「死亡」。
解析計画
(主要評価期のみについて記載)
(1)有効性の評価項目
- 1)主要評価項目:6分間歩行距離の変化量および血行動態パラメータ(CI、PAPm、PVRI)の変化量について中央値[25%点-75%点]を算出した。
- 2)副次評価項目:修正ボルグスケールの変化量、EQ-5Dスコア、主要評価項目以外の血行動態パラメータおよびPAH症状の改善スコアの変化量について、中央値[25%点-75%点]を算出した。また、WHO機能分類の変化、PAH症状の変化について割合を算出した。
- 3)欠測データの補完方法:6分間歩行距離、修正ボルグスケールは、臨床的悪化により未実施の場合は、それぞれ0 m、10に補完した。それ以外の理由により未実施の場合は、直前のデータをLOCFで補完した。 PAH症状改善スコアは、中止の理由が臨床的悪化の場合は、-5点に補完し、それ以外の理由により中止の場合は、投与後のデータを対象にLOCFで補完した。
(2)安全性の評価:有害事象の発現率を評価した。