- 医療関係者向けホーム
- 消化器領域
- リアルダ®錠
- Clinical Study:成人(各臨床試験での定義:16歳以上)の潰瘍性大腸炎患者に対する臨床成績:活動期 試験の概要
 Clinical Study
Clinical Study
成人(各臨床試験での定義:16歳以上)の潰瘍性大腸炎患者に対する臨床成績:活動期
活動期:pH依存型メサラジン放出調節製剤 3600mg/日(1日3回)を対照とした活動期における国内第Ⅲ相試験〔非劣性試験〕
-
[持田製薬社内資料:国内第Ⅲ相試験 ―メサラジンを対照とした軽症~中等症の活動期の潰瘍性大腸炎における有効性及び安全性の検討―
(2016年9月28日承認、CTD 2.7.6.18)]〈承認時評価資料〉
試験の概要
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
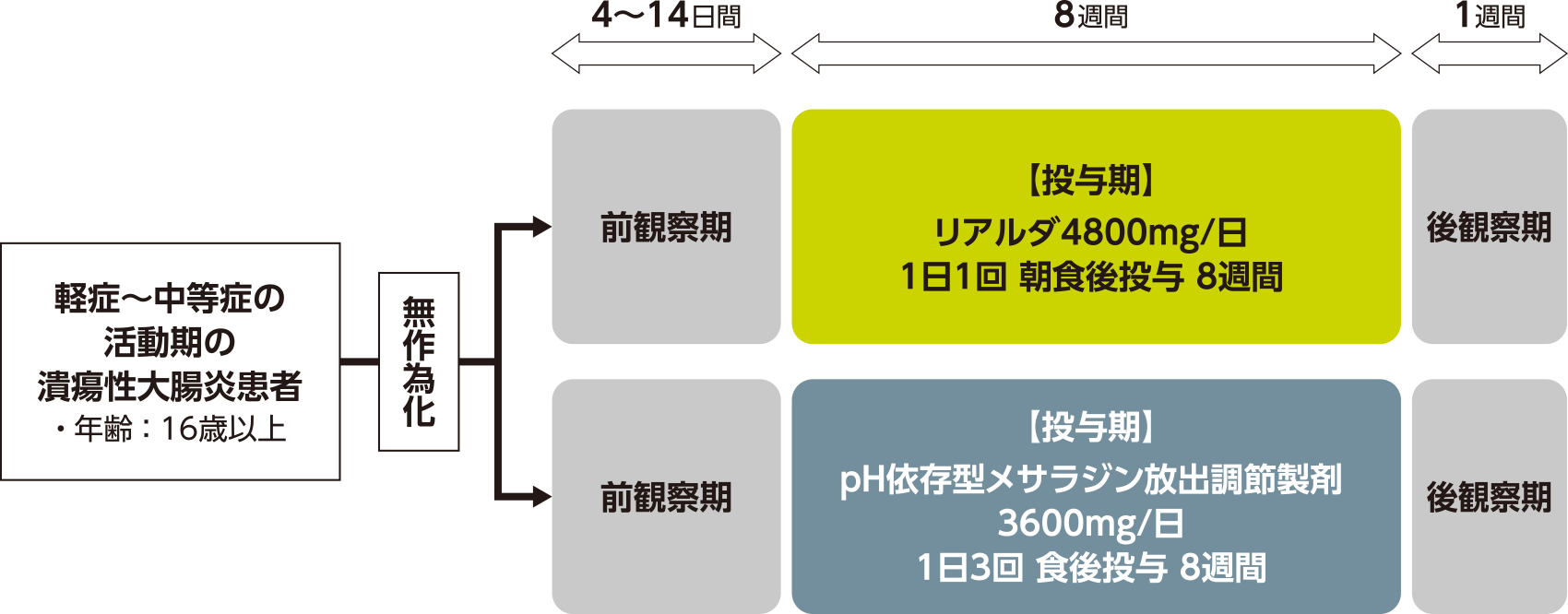
試験デザイン
多施設共同・ランダム化・二重盲検・実薬対照・並行群間・第Ⅲ相試験(ダブルダミー法)
目的
軽症~中等症の活動期の潰瘍性大腸炎患者を対象に、pH依存型メサラジン放出調節製剤3600mg/日(1日3回)に対するリアルダ4800mg/日(1日1回)の「非劣性」を検証することを目的とした。
対象
軽症~中等症の活動期の潰瘍性大腸炎患者280例
〔FAS(Full Analysis Set):278例、PPS(Per Protocol Set):267例、安全性解析対象集団:280例〕
| 【選択基準】 |
|
|---|
方法
対象患者をランダムにリアルダ4800mg/日(1日1回)群、pH依存型メサラジン放出調節製剤3600mg/日(1日3回)群の2群に割り付けた。試験期間は、前観察期(4~14日間)、投与期(8週間)、後観察期(1週間)から構成された。
主要評価項目
| 〈有効性〉 | UC-DAIスコアの変化量(投与期終了時 − 投与期開始時)
|
|---|---|
| 〈安全性〉 | 有害事象(投与期) |
副次評価項目
| 〈有効性〉 | UC-DAIを構成する各スコアの推移、寛解率、臨床的寛解率、内視鏡的寛解率、改善率 |
|---|---|
| 〈安全性〉 | 有害事象(前観察期、後観察期)および副作用(投与期、後観察期) |

解析計画
主要評価項目は、閉手順を用いて階層的(①→②)に解析した。UC-DAIスコアの変化量に関して、まず①としてPPSを解析対象に共分散分析を行い、投与群間の平均値の差(リアルダ4800mg/日群の変化量-pH依存型メサラジン放出調節製剤3600mg/日群の変化量)の両側95%信頼区間の上限が、治験実施計画書で規定した非劣性限界値「1.1」を下回った場合に「非劣性」が検証されることとした。共分散分析モデルは、群を主効果、投与期開始時のUC-DAIスコアを共変量とすることとした。「非劣性」が検証された場合に限り、②としてFASを解析対象に投与群間の平均値の差の両側95%信頼区間に基づき、「優越性」の確認も行うこととした。
なお、サブグループ解析として、UC-DAIスコアの変化量を「病変の拡がり注)別」に検討した。
「投与期終了時」の評価は、投与期8週時または中止時のスコアを用いることとした。
「臨床的寛解」について、欠測値はLOCF(Last Observation Carried Forward)法により補完し、補完できない場合は「非臨床的寛解」として取り扱った。「寛解」、「内視鏡的寛解」および「改善」について、欠測値は「非寛解」、「非内視鏡的寛解」および「非改善」として取り扱った。
- 注)臨床試験時は「病変範囲」としましたが、『潰瘍性大腸炎・クローン病 診断基準・治療指針』に則り、
「病変の拡がり」と表現を変更しています。
■UC-DAI(Ulcerative Colitis Disease Activity Index)スコア:4つの評価項目をそれぞれ4段階(0~3)でスコア化して、合計(0~12)。
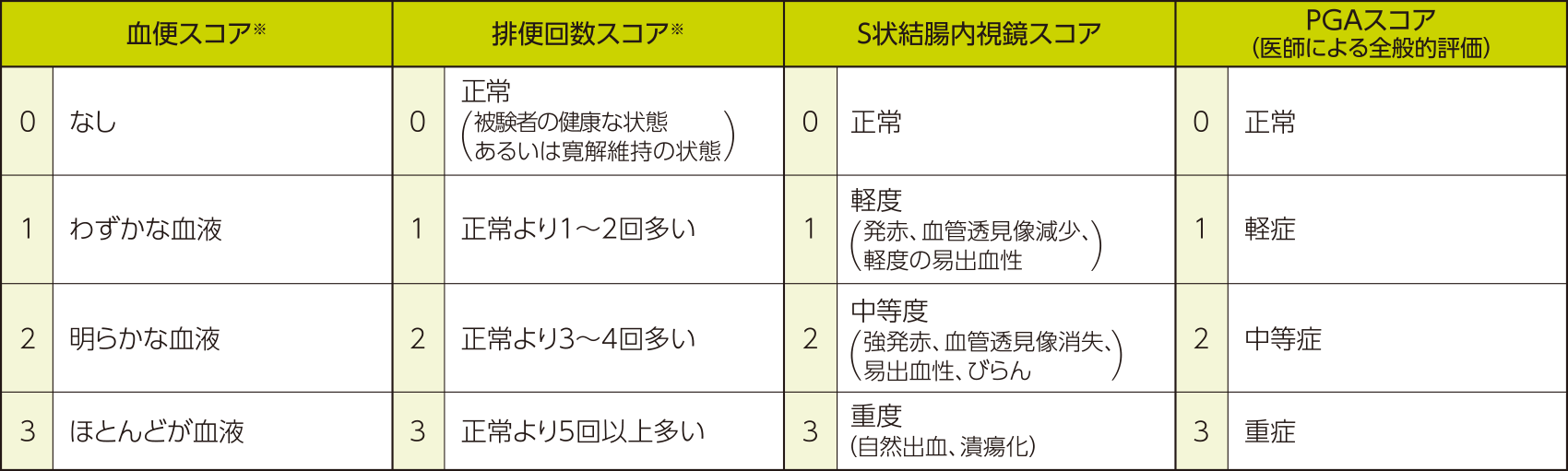
- ※:血便スコアと排便回数スコアについては、3日間のスコアの平均値(小数点第一位を四捨五入)を用いることとした。
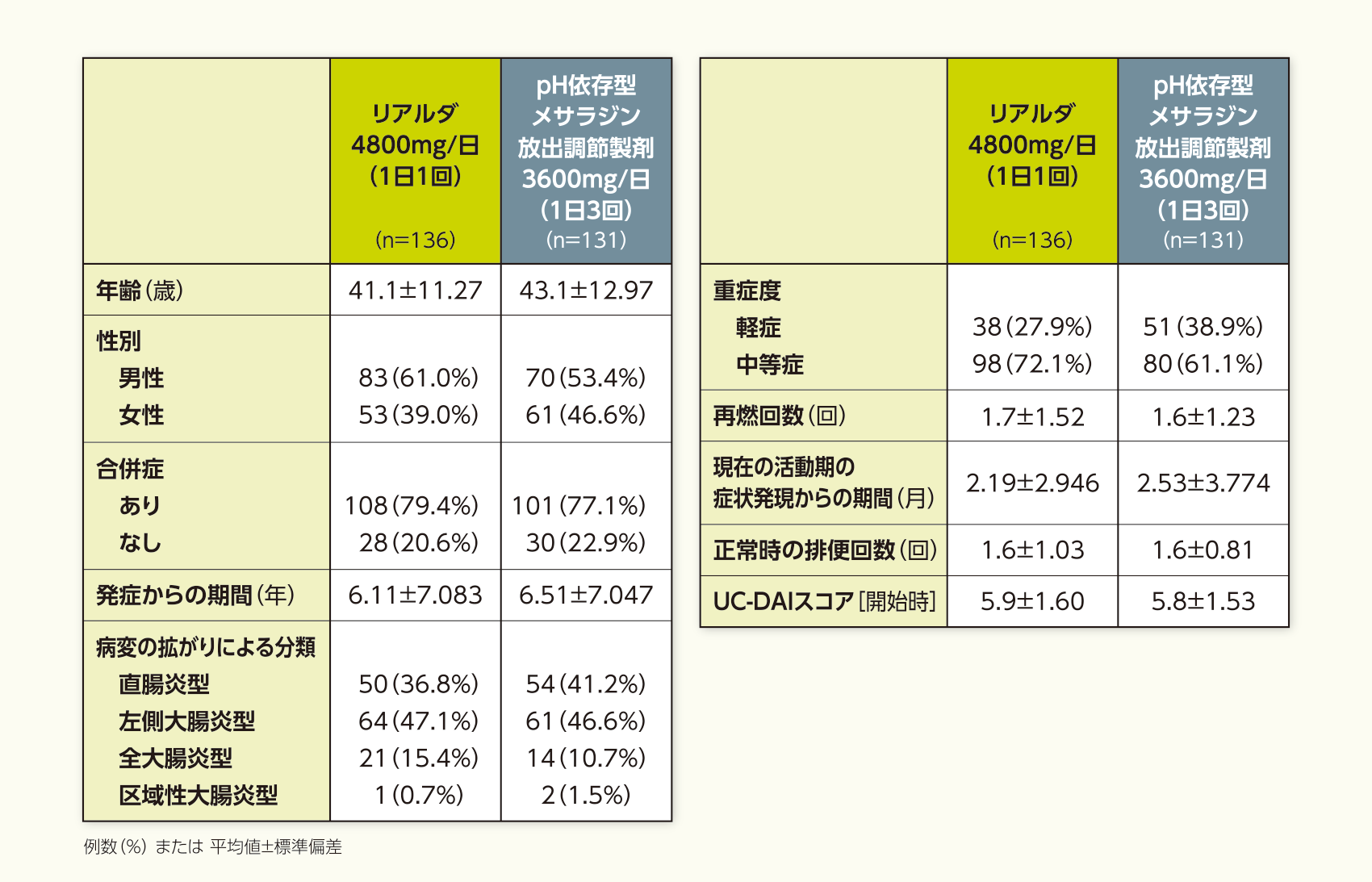
患者背景(PPS)
2025年12月作成
14407-18/N14 B8 MDC

