- 医療関係者向けホーム
- 消化器領域
- リアルダ®錠
- Clinical Study:成人(各臨床試験での定義:16歳以上)の潰瘍性大腸炎患者に対する臨床成績:寛解期 試験の概要
 Clinical Study
Clinical Study
成人(各臨床試験での定義:16歳以上)の潰瘍性大腸炎患者に対する臨床成績:寛解期
寛解期:時間依存型メサラジン放出調節製剤2250mg/日(1日3回)を対照とした寛解期における国内第Ⅲ相試験〔非劣性試験〕
-
[持田製薬社内資料:国内第Ⅲ相試験 ―メサラジンを対照とした寛解期の潰瘍性大腸炎における有効性及び安全性の検討―
(2016年9月28日承認、CTD 2.7.6.17)]〈承認時評価資料〉
試験の概要
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
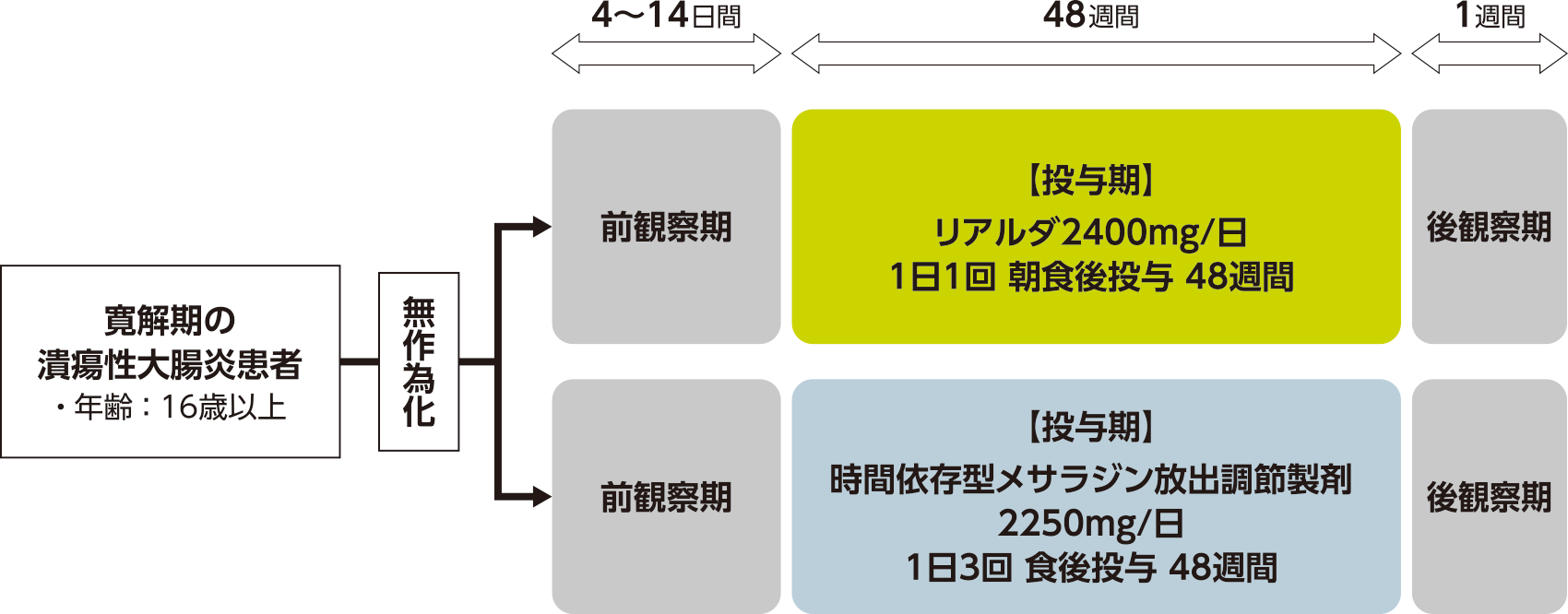
試験デザイン
多施設共同・ランダム化・二重盲検・実薬対照・並行群間・第Ⅲ相試験(ダブルダミー法)
目的
寛解期の潰瘍性大腸炎患者を対象に、時間依存型メサラジン放出調節製剤2250mg/日(1日3回)に対するリアルダ2400mg/日(1日1回)の「非劣性」を検証することを目的とした。
対象
寛解期の潰瘍性大腸炎患者203例
〔PPS(Per Protocol Set):199例、安全性解析対象集団:203例〕
| 【選択基準】 |
|
|---|
方法
対象患者をランダムにリアルダ2400mg/日(1日1回)群、時間依存型メサラジン放出調節製剤2250mg/日(1日3回)群の2群に割り付けた。試験期間は、前観察期(4~14日間)、投与期(48週間)、後観察期(1週間)から構成された。
主要評価項目
| 〈有効性〉 | 血便の非発現率:投与期(2次登録日~投与期終了時)に血便スコアが0であった患者の割合
|
|---|---|
| 〈安全性〉 | 有害事象(投与期) |
副次評価項目
| 〈有効性〉 | 血便の非発現期間、再燃率 など
|
|---|---|
| 〈安全性〉 | 有害事象(前観察期、後観察期)および副作用(投与期、後観察期) |
UC-DAIスコアの概要については、こちらをご参照ください。
サブグループ解析
潰瘍性大腸炎の臨床経過注)別にみた血便の非発現率
解析計画
投与期48週時の血便の非発現率に関してPPSを解析対象とし、投与群間の血便の非発現率の差(リアルダ2400mg/日群-時間依存型メサラジン放出調節製剤2250mg/日群)の両側95%信頼区間の下限が、治験実施計画書で規定した非劣性限界値「-10%」を上回った場合に「非劣性」が検証されることとした。
なお、サブグループ解析として、血便の非発現率を「潰瘍性大腸炎の臨床経過注)別」に比較した。
「投与期終了時」の評価は、投与期48週時または中止時のスコアを用いることとした。
欠測の取り扱いについて、有効性評価指標の評価データが欠測しデータを代入する場合は、LOCF(Last Observation Carried Forward)法による補完を実施した。再燃の評価欠測例において、成績を補完する場合は「再燃あり」として補完した。
- 注)臨床試験時は「病型」としましたが、『潰瘍性大腸炎・クローン病 診断基準・治療指針』に則り、「臨床経過」と表現を変更しています。
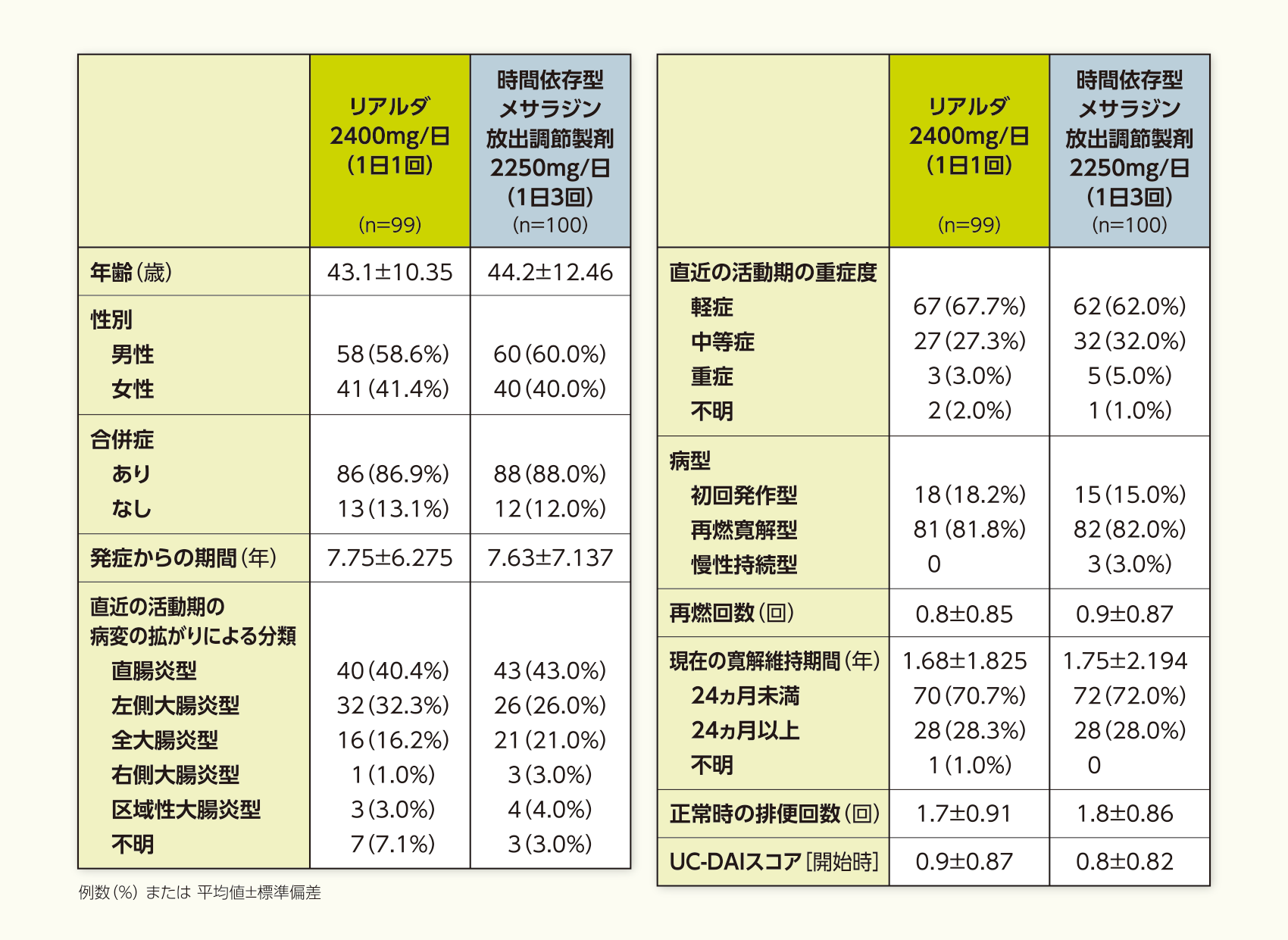
患者背景(PPS)
2025年12月作成
14407-18/N14 B8 MDC

