- 医療関係者向けホーム
- 消化器領域
- オンボー®
- Clinical Study(潰瘍性大腸炎)「寛解導入期」に対する効果 試験の概要
 Clinical Study(潰瘍性大腸炎)「寛解導入期」に対する効果
Clinical Study(潰瘍性大腸炎)「寛解導入期」に対する効果
「寛解導入期」に対する効果
第Ⅲ相無作為化比較試験:LUCENT-1(AMAN)試験(国際共同試験)1)
- 1)社内資料:潰瘍性大腸炎患者を対象とした国際共同第Ⅲ相試験(AMAN試験、LUCENT-1:寛解導入療法)(承認時評価資料)
- 2)D'Haens, G. et al.:N Engl J Med., 388(26), 2444(2023)
本研究はイーライリリーの支援により行われた。本論文の著者のうち8名は、イーライリリーの社員である。 - 3)Kobayashi, T. et al.:Intest Res., 22(2), 172(2024)
Reprinted from Intest Res 2024;22:172 with permission
本研究はイーライリリーの支援により行われた。本論文の著者のうち8名は、イーライリリーの社員である。著者にイーライリリーより
コンサルタント料等を受領している者が含まれる。著者に持田製薬より講演料、研究助成金等を受領している者が含まれる。
試験の概要
「警告・禁忌を含む注意事項等情報」等については電子添文をご参照ください。
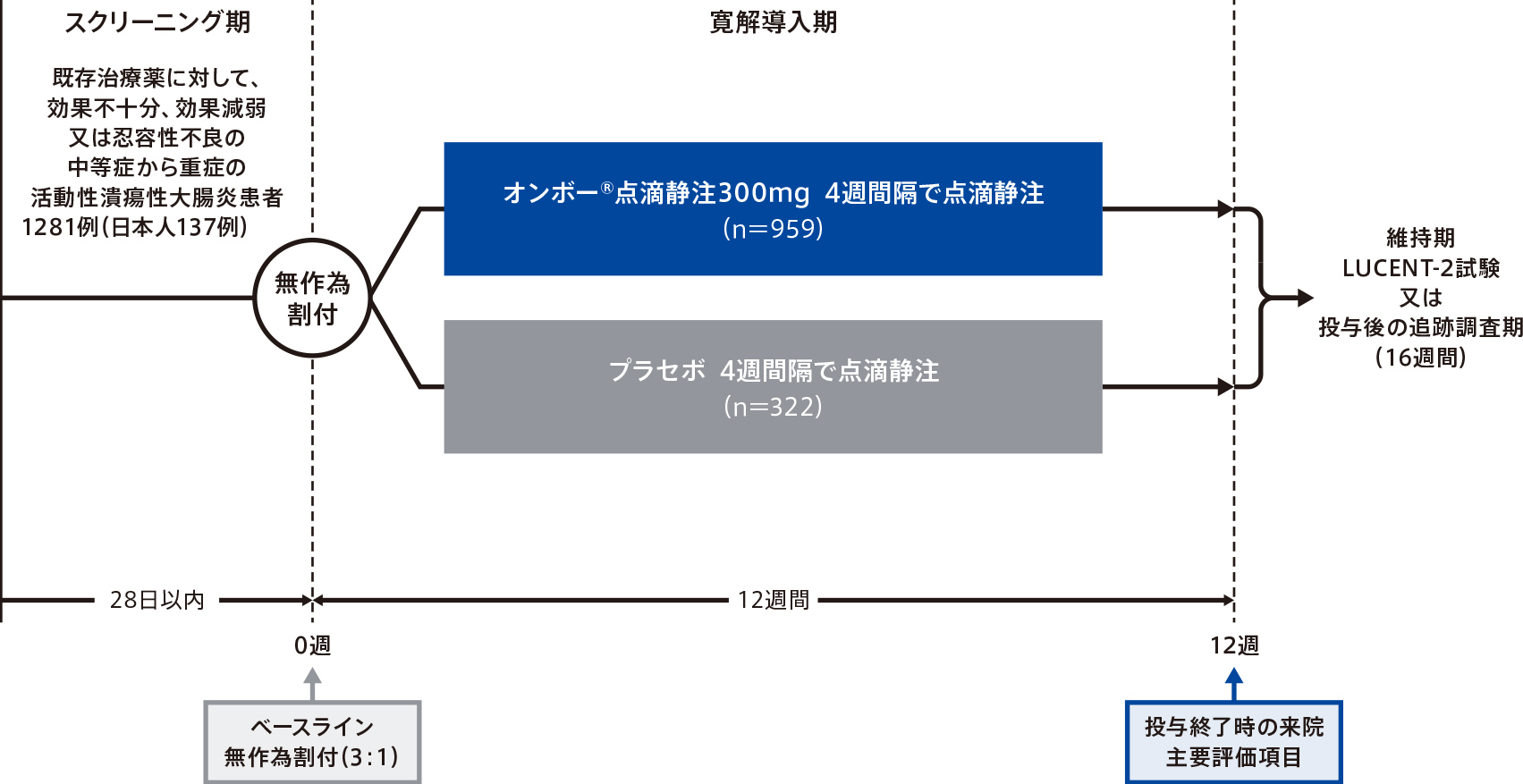
試験デザイン
多施設共同注1)、無作為化、二重盲検、プラセボ対照、並行群間比較検証試験
- 注1)471施設(35ヵ国)
目的
中等症から重症の潰瘍性大腸炎患者を対象として、12週時点でオンボー®がプラセボより臨床的寛解の導入で優れているという仮説を検証する。
対象
既存治療薬〔ステロイド系薬剤、免疫調節薬、生物学的製剤(抗TNFα抗体又は抗α4β7インテグリン抗体)又はJAK阻害薬(トファシチニブ)のうち少なくとも1剤〕に対して、効果不十分、効果減弱又は忍容性不良の中等症から重症の活動性潰瘍性大腸炎患者注2)1281例(日本人137例を含む)
- 注2)スクリーニング(ベースライン来院前の最長28日間)時に18歳以上80歳以下、ベースラインで潰瘍性大腸炎の診断歴を3ヵ月以上有する、潰瘍性大腸炎の内視鏡所見を有する、潰瘍性大腸炎の診断を裏付ける病理組織検査報告書を有する、ベースラインの前14日以内の内視鏡検査でmodified Mayo score(MMS)が4~9かつ内視鏡所見サブスコアが2以上、潰瘍性大腸炎の所見が直腸から口側の結腸に進展している、従来の治療薬(ステロイド系薬剤又は免疫調節薬)、生物学的製剤(抗TNFα抗体又は抗α4β7インテグリン抗体)又はJAK阻害薬(トファシチニブ)に対する治療反応が不良
投与方法
患者を生物学的製剤に対する治療反応性不良の状況(あり/なし)、ベースラインのステロイド系薬剤の使用(あり/なし)、ベースラインの疾患活動性(MMS 4~6/7~9)、及び地域(北米/欧州/その他)に基づいて層別化し、オンボー®点滴静注300mg群又はプラセボ群に3:1の比で無作為に割り付け、オンボー®点滴静注300mg又はプラセボを0週(投与開始)、4週、8週の3回点滴静注した。
評価項目
有効性
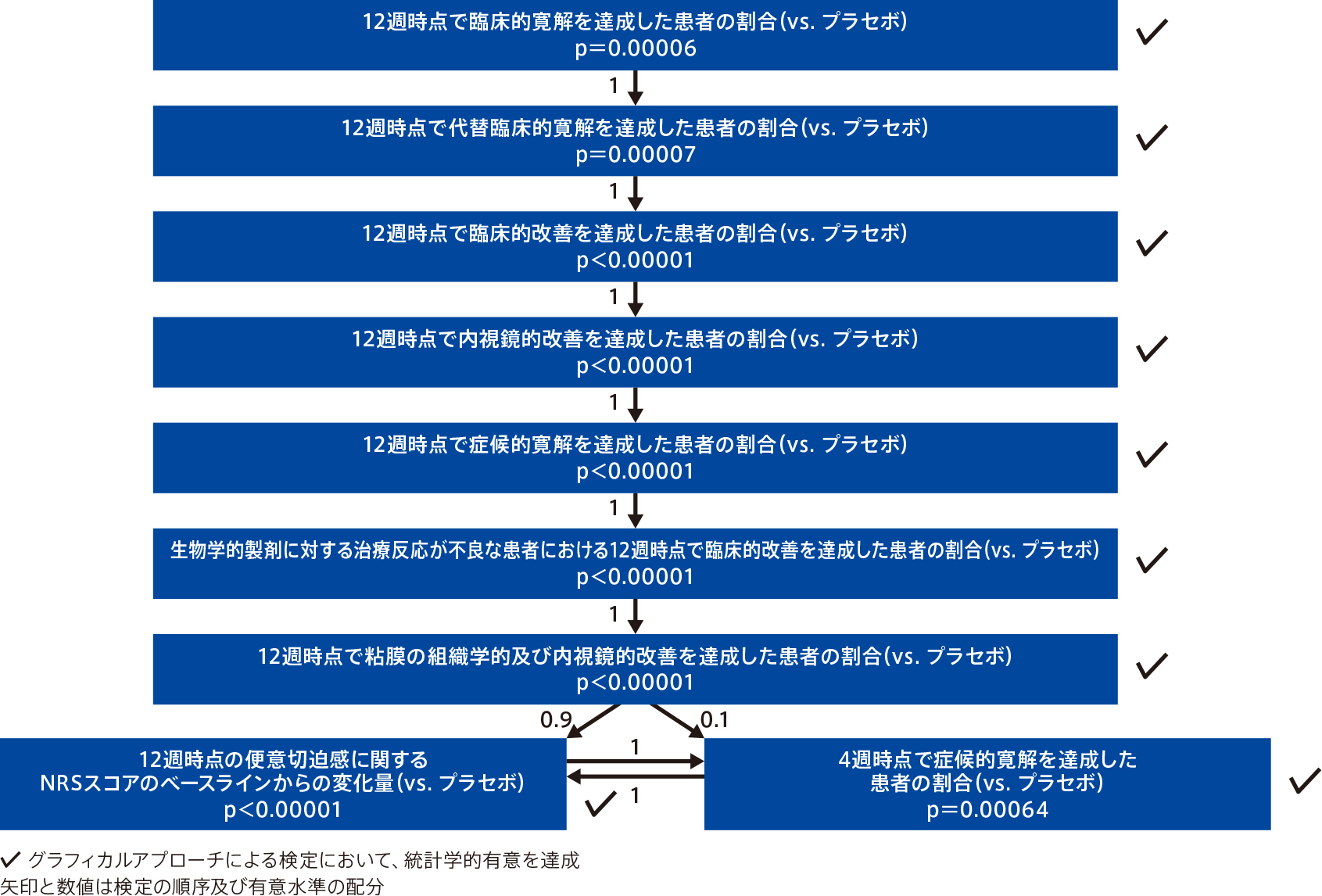
<主要評価項目:検証的解析項目>
- 12週時点で臨床的寛解を達成した患者の割合
<重要な副次評価項目>
- 12週時点で代替臨床的寛解を達成した患者の割合
- 12週時点で臨床的改善を達成した患者の割合
- 12週時点で内視鏡的改善を達成した患者の割合
- 12週時点で症候的寛解を達成した患者の割合
- 生物学的製剤に対する治療反応が不良な患者における12週時点で臨床的改善を達成した患者の割合(サブグループ解析)
- 12週時点で粘膜の組織学的及び内視鏡的改善を達成した患者の割合
- 12週時点の便意切迫感に関するNRSスコア注3)のベースラインからの変化量
- 4週時点で症候的寛解を達成した患者の割合
<その他の副次評価項目>
- 2、6、8週時点で症候的寛解を達成した患者の割合
- 2、4、8、12週時点の直腸出血サブスコア及び排便回数サブスコアのベースラインからの変化量
- 2、4、8週時点の便意切迫感に関するNRSスコア注3)のベースラインからの変化量
- ベースラインで便意切迫感に関するNRSスコア注3)が3以上の患者における12週時点までの便意切迫感に関するNRSスコア0又は1を達成した患者の割合
- ベースラインで便意切迫感に関するNRSスコア注3)が3以上の患者における12週時点までの便意切迫感に関するNRSスコアがベースラインから3ポイント以上改善した患者の割合
など
薬物動態
血清中ミリキズマブ濃度
安全性
有害事象、免疫原性 など
- 注3)過去24時間における患者の排便に対する切迫感(突然又は即時の必要性)の重症度を0(便意切迫感なし)から10(考えられる最も悪い便意切迫感)の11ポイントの範囲で評価する患者報告アウトカムで、連日電子日誌に記録された7日間のデータを平均して算出
評価項目の用語の定義

| MMSとMayoスコア: | Mayoスコアは潰瘍性大腸炎の重症度を評価するための臨床的及び内視鏡的指標を組み合わせたもの。4つのサブスコア(排便回数サブスコア、直腸出血サブスコア、内視鏡所見サブスコア、医師による全般的評価)で構成される。各サブスコアは0~3の範囲であり、Mayoスコアの合計は0~12の範囲となる。MMSは、排便回数サブスコア、直腸出血サブスコア、及び内視鏡所見サブスコアを合計して算出され、0~9の範囲となる。 |
|---|---|
| Geboesスコア: | 組織学的活動性評価の指標。 |
解析計画
有効性の解析はmITT解析対象集団注4)、安全性の解析は安全性解析対象集団注5)を対象として実施した。主要評価項目及び重要な副次評価項目では、事前に規定したグラフィカルアプローチによる多重検定法を用いて、両側有意水準0.00125で全体の第1種の過誤の確率を制御し、両側99.875%信頼区間を示した。
主要評価項目などの二値変数の有効性評価項目の解析では、Cochran-Mantel-Haenszel(CMH)検定を実施した。CMH検定では、生物学的製剤に対する治療反応性不良の状況(あり/なし)、ベースラインのステロイド系薬剤の使用(あり/なし)、ベースラインの疾患活動性(MMS 4~6/7~9)、地域(北米/欧州/その他)を共変量の調整因子とした。便意切迫感に関するNRSスコアのベースラインからの変化量などの連続量は、事前に規定された時点を含む繰り返し測定値に関する混合効果モデル(mixed-effects model for repeated measures:MMRM)を用いて解析した。MMRMでは、投与群、ベースライン値、時期、ベースライン値と時期の交互作用、投与群と時期の交互作用、及びCMH検定で使用した因子と同じ共変量をモデルに含めた。
欠測データは、二値変数の有効性評価項目の解析では、non-responder imputation(NRI)法で補完した。患者が臨床的改善の基準を満たしていない場合、評価時点で臨床的改善データが欠測している場合、その患者をノンレスポンダーとみなした。連続量では、ランダムな欠測を仮定したMMRMを主要解析に用いた。連続変数の欠測値は、単一の時点でのデータ欠損の場合はmBOCF法、有害事象で投与を中止した場合はベースラインの値、有害事象以外の理由で投与を中止した場合は得られた最終観測データを用いて補完した。
二値変数の有効性評価項目の投与群比較は、共通リスク差(群間差)の推定値を使用し、両側信頼区間も記載した。連続量の有効性評価項目の投与群比較は、MMRMを使用した。投与群間の統計的比較には、最小二乗平均値のType Ⅲ平方和を用いた。特に規定しない限り、最小二乗平均値の差、標準誤差、p値、及び95%信頼区間を記載した。
なお、有効性評価項目について、サブグループ解析注6)を行うことを事前に規定した。重要な副次評価項目及びその他の副次評価項目である症候的寛解を達成した患者の割合、その他の副次評価項目である便意切迫感に関するNRSスコア0又は1を達成した患者の割合及び便意切迫感に関するNRSスコアがベースラインから3ポイント以上改善した患者の割合の評価期間は、1週から12週まで1週ごととした。
免疫原性がミリキズマブの有効性、安全性及び薬物動態へ及ぼす影響を評価した。
- 注4)無作為割付されて治験薬の投与を1回以上受けたすべての患者(患者が正しい投与を受けたか、又は治験実施計画書を遵守したかを問わない)。ポーランド及びトルコで発生した電子臨床アウトカム評価(electronic clinical outcome assessment:eCOA)エラーの影響を受けた患者は除外した。
- 注5)無作為割付されて治験薬の投与を1回以上受けたすべての患者(患者が正しい投与を受けたか、又は治験実施計画書を遵守したかを問わない)。
- 注6)以下の部分集団で解析を実施
・日本人
・生物学的製剤及びJAK阻害薬の使用歴がない患者(BIO・JAK naive)
・1種類以上の生物学的製剤又はJAK阻害薬に対する治療反応が不良な患者(BIO・JAK failure) - 注6)以下の部分集団で解析を実施