- 医療関係者向けホーム
- 消化器領域
- オンボー®
- Clinical Study(クローン病)「クローン病」に対する効果 試験の概要
 Clinical Study(クローン病)「クローン病」に対する効果
Clinical Study(クローン病)「クローン病」に対する効果
「クローン病」に対する効果
第Ⅲ相無作為化比較試験:VIVID-1(AMAM)試験(国際共同試験)1、2)
- 1)社内資料:クローン病患者を対象とした国際共同第Ⅲ相試験(AMAM試験、VIVID-1)(承認時評価資料)
- 2)Ferrante, M. et al.:Lancet., 404(10470), 2423(2024)
- 本研究はイーライリリーの支援により行われた。本論文の著者のうち9名は、イーライリリーの社員である。著者にイーライリリーより講演料、
コンサルタント料等を受領している者が含まれる。著者に持田製薬より講演料、研究助成金等を受領している者が含まれる。
試験の概要
「警告・禁忌を含む注意事項等情報」等については電子添文をご参照ください。
試験デザイン
多施設共同注1)、無作為化、二重盲検、ダブルダミー、プラセボ及び実薬対照、treat-through 、並行群間比較検証試験
- 注1)328施設(33ヵ国)

- 注2)8~20週まで、すべての患者に割り付けられた治療及び対応するプラセボの点滴静注及び皮下投与の両方を実施した。
- 注3)無作為割付された1152例のうち、1150例の患者が1回以上の治験薬投与を受けた(mITT集団)。mITT集団(1150例)のうち1065例(日本人26例)は、ベースライン時のSES-CDスコアが7以上(回腸限局型クローン病を有する患者では4以上)の患者であった(PAS集団)。
- 注4)本試験では、ウステキヌマブの国内承認外の用法及び用量が用いられた。
- 注5)排便回数、腹痛スコアのいずれもベースラインから30%以上の減少が認められない、又はいずれかがベースラインから悪化している患者をノンレスポンダーと定義した。
目的
中等症から重症の活動性クローン病患者を対象として、以下の有効性評価項目を指標としたプラセボに対するオンボー®の優越性を評価する。
- ・PROによる臨床的改善(12週)かつ内視鏡的改善(52週)
- ・PROによる臨床的改善(12週)かつCDAIによる臨床的寛解(52週)
対象
既存治療薬〔ステロイド系薬剤、免疫調節薬又は生物学的製剤(抗TNFα抗体又は抗α4β7インテグリン抗体)のうち少なくとも1剤〕に対して、効果不十分、効果減弱又は不耐の中等症から重症の活動性クローン病患者注6)1152例(日本人28例を含む)
- 注6)スクリーニング時に18歳以上80歳以下の男性又は女性で以下に該当する患者
- ・組入れの3ヵ月以上前にクローン病又は瘻孔形成型クローン病と診断され、臨床的、内視鏡的、及び組織学的基準により診断が確定している
- ・ベースライン時点で、1日の平均排便回数(重みづけなし)が4回以上(Bristol stool scale categoryで6又は7と定義される軟便及び水様便)及び/又は1日の平均腹痛スコア(重みづけなし)が2以上と定義される、中等症から重症の活動性クローン病を有する
- ・無作為化前21日以内に、SES-CDスコア(中央判定)が7以上の回腸結腸型クローン病又は4以上の回腸限局型クローン病を有する*1
- ・1種類以上の治療薬[ステロイド系薬剤、免疫調節薬〔アザチオプリン、6-メルカプトプリン(クローン病に対して国内未承認)、メトトレキサート(クローン病に対して国内未承認)、及びチオプリン製剤とアロプリノール(クローン病に対して国内未承認)併用〕、クローン病の治療薬として承認された生物学的製剤(抗TNFα抗体又は抗α4β7インテグリン抗体)]で治療不良(効果不十分*2、効果減弱又は不耐)
- *1 SES-CDスコアが3以上7未満(回腸限局型クローン病を有する患者ではSES-CDスコア4未満)で、(回腸又は結腸、もしくはその両方に)一つ以上の大きな潰瘍があるため「潰瘍のサイズ」の項目でスコアが2以上、「潰瘍の面積」の項目でスコアが1以上になる患者も組入れ可とした。ただし、総組入れ患者数の約10%に制限した。
- *2 既存治療薬効果不十分の定義(プロトコール登録基準[8a])に該当する薬剤のうち、セルトリズマブペゴル、ナタリズマブはクローン病に対して国内未承認。
投与方法
第1期(0~12週)注7)
以下の投与群に6:3:2の割合で無作為割付した注8)。
- ・オンボー®群:0、4、及び8週時にオンボー®900mgを4週間隔で点滴静注した。
- ・ウステキヌマブ群:0週時にウステキヌマブ6mg/kgを点滴静注、及び8週時にウステキヌマブ90mgを皮下投与した注9)。
- ・プラセボ群:プラセボを点滴静注及び皮下投与した。
第2期(12~52週)注7)
- ・オンボー®群:オンボー®300mgを4週間隔で皮下投与した。
- ・ウステキヌマブ群:ウステキヌマブ90mgを16週時、それ以降は8週間隔で皮下投与した注9)。
- ・プラセボ群(第1期の12週時点のレスポンダー注10)):12週から20週の間はプラセボの点滴静注及び皮下投与を継続し、24週から52週はプラセボを皮下投与した。
- ・プラセボ群(第1期の12週時点のノンレスポンダー注10)):オンボー®900mgを4週間隔で3回点滴静注し、その後はオンボー®300mgを4週間隔で皮下投与した。
- 注7)盲検性維持のため、ダブルダミー法を用いて治験薬の投与経路及び投与頻度の違いを盲検化した。
- 注8)層別因子:生物学的製剤の治療不良歴(あり/なし)、ベースラインのステロイド系薬剤使用(あり/なし)、ベースラインのSES-CDスコア(12未満/12以上)、地域(北米/欧州/その他)、指示変数(ベースラインの排便回数が7回以上又はベースラインの腹痛スコアが2.5以上)。
- 注9)本試験では、ウステキヌマブの国内承認外の用法及び用量が用いられた。
- 注10)排便回数、腹痛スコアのいずれもベースラインから30%以上の減少が認められない、又はいずれかがベースラインから悪化している患者をノンレスポンダーと定義した。
オンボー®点滴静注の用法及び用量に関連する注意(抜粋)
〈効能共通〉
- 7.1本剤と他の生物製剤又はヤヌスキナーゼ(JAK)阻害剤との併用について安全性及び有効性は確立していないので併用を避けること。
〈クローン病〉
- 7.4本剤の3回目投与の4週後にミリキズマブ(遺伝子組換え)皮下投与用製剤の投与を開始すること(皮下投与用製剤による治療の用法及び用量は、ミリキズマブ(遺伝子組換え)皮下投与用製剤の電子添文を参照すること)。本剤による治療開始から24週後までに効果が得られない場合には、投与継続の必要性を検討すること。
- 7.5ミリキズマブ(遺伝子組換え)皮下投与用製剤による治療中に効果が減弱し、再度の本剤の3回投与により治療効果が得られた場合には、3回目投与の4週後から皮下投与用製剤の投与を再開すること。再度の本剤の3回投与の4週後に治療効果が得られない場合は、本剤の投与を中止し、他の治療法への切替えを考慮すること。また、皮下投与用製剤による治療中に再び効果が減弱した場合には、他の治療法への切替えを考慮すること。皮下投与用製剤による治療中の2回目以降の効果減弱時に、本剤を投与した場合の安全性及び有効性を評価する臨床試験は実施していない。
オンボー®皮下注の用法及び用量に関連する注意(抜粋)
〈効能共通〉
- 7.1本剤と他の生物製剤又はヤヌスキナーゼ(JAK)阻害剤との併用について安全性及び有効性は確立していないので併用を避けること。
〈クローン病〉
- 7.3ミリキズマブ(遺伝子組換え)治療開始から24週後までに効果が得られない場合には、投与継続の必要性を検討すること。
- 7.4本剤の皮下投与による治療中に効果が減弱し、再度のミリキズマブ(遺伝子組換え)点滴静注製剤の3回投与により治療効果が得られた場合には、3回目投与の4週後から本剤の皮下投与を再開すること(効果減弱時における用法及び用量は、ミリキズマブ(遺伝子組換え)点滴静注製剤の電子添文を参照すること)。また、本剤の皮下投与による治療中に再び効果が減弱した場合には、他の治療法への切替えを考慮すること。本剤の皮下投与による治療中の2回目以降の効果減弱時に、ミリキズマブ(遺伝子組換え)点滴静注製剤を投与した場合の安全性及び有効性を評価する臨床試験は実施していない。

ウステキヌマブ皮下注のクローン病に対しての国内で承認された用法及び用量
ウステキヌマブ(遺伝子組換え)の点滴静注製剤を投与8週後に、通常、成人にはウステキヌマブ(遺伝子組換え)として90mgを皮下投与し、以降は12週間隔で90mgを皮下投与する。なお、効果が減弱した場合には、投与間隔を8週間に短縮できる。
評価項目
有効性
●プラセボに対するオンボー®の優越性を評価
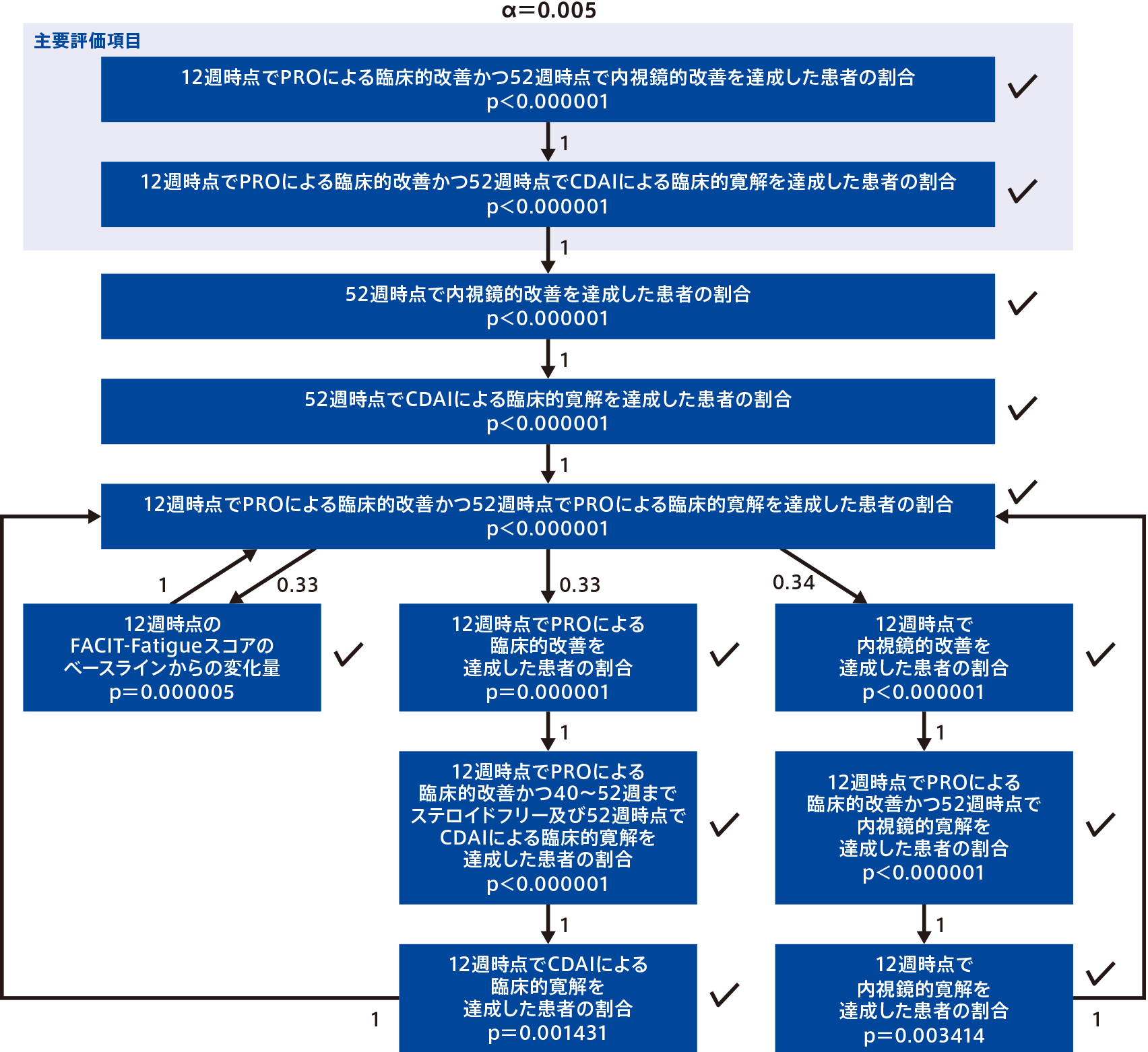
<主要評価項目:検証的解析項目※>
- 12週時点でPROによる臨床的改善かつ52週時点で内視鏡的改善を達成した患者の割合
- 12週時点でPROによる臨床的改善かつ52週時点でCDAIによる臨床的寛解を達成した患者の割合
- ※2つの主要評価項目をそれぞれ評価し、両者とも達成した場合に、有効性が検証されたと判断する
<重要な副次評価項目>
- 12週時点で内視鏡的改善を達成した患者の割合
- 52週時点で内視鏡的改善を達成した患者の割合
- 12週時点でCDAIによる臨床的寛解を達成した患者の割合
- 52週時点でCDAIによる臨床的寛解を達成した患者の割合
- 12週時点でPROによる臨床的改善を達成した患者の割合
- 12週時点でPROによる臨床的改善かつ52週時点で内視鏡的寛解を達成した患者の割合
- 12週時点で内視鏡的寛解を達成した患者の割合
- 12週時点のFACIT-Fatigueスコアのベースラインからの変化量
- 12週時点でPROによる臨床的改善かつ52週時点でPROによる臨床的寛解を達成した患者の割合
- 12週時点でPROによる臨床的改善かつ40~52週までステロイドフリー及び52週時点でCDAIによる臨床的寛解を達成した患者の割合
<その他の副次評価項目>
- 0週から52週の各来院時のCDAIによる臨床的寛解を達成した患者の割合
- 0週から52週の各来院時のPROによる臨床的改善を達成した患者の割合
- 12週時点の便意切迫感に関するNRSスコア注11)のベースラインからの変化量
- 52週時点の便意切迫感に関するNRSスコア注11)のベースラインからの変化量
- 0週から52週の各来院時の便意切迫感に関するNRSスコア注11)のベースラインからの変化量
- ベースラインで便意切迫感に関するNRSスコア注11)が3以上の患者における12週時点の便意切迫感に関するNRSスコアが2以下を達成した患者の割合
- ベースラインで便意切迫感に関するNRSスコア注11)が3以上の患者における12週時点でPROによる臨床的改善かつ52週時点の便意切迫感に関するNRSスコアが2以下を達成した患者の割合
など
●ウステキヌマブに対するオンボー®の非劣性又は優越性を評価
<重要な副次評価項目>
- 52週時点でCDAIによる臨床的寛解を達成した患者の割合(非劣性)
- 52週時点で内視鏡的改善を達成した患者の割合(優越性)
<その他の副次評価項目>
- 52週時点で内視鏡的改善かつCDAIによる臨床的寛解を達成した患者の割合
安全性
有害事象、免疫原性(抗ミリキズマブ抗体の発現状況)など
- 注11)過去24時間における患者の排便に対する切迫感(突然又は即時の必要性)の重症度を0(便意切迫感なし)から10(考えられる最も悪い便意切迫感)の11ポイントの範囲で評価する患者報告アウトカムで、電子日誌に記録された来院前の12日間のうち直近の7日間のデータを平均して算出
評価項目の用語の定義

| PRO: | CDAIの排便回数(stool frequency:SF)及び腹痛スコア(abdominal pain:AP)の2項目で定義した患者報告アウトカム(patient-reported outcome, 2 of the patient-reported items of the CDAI[SF and AP])。CDAIの2つの患者報告項目に基づいている(重みづけしていない1日の平均値)。
|
|---|
| SES-CD: | Simple endoscopic score for Crohn‘s disease. 回腸結腸の5つのセグメント(回腸、右側結腸、横行結腸、左側結腸、直腸)で内視鏡により評価する4つの項目(潰瘍の有無とサイズ、潰瘍の面積の割合、その他の病変の面積の割合、及び狭窄の有無と重症度)に基づいている。回腸結腸の5つのセグメントについて、内視鏡により評価する上記4項目をそれぞれ0~3で点数化し、SES-CDの合計スコアは0~56の範囲となる。 |
|---|
| CDAI: | クローン病活動指数(Crohn‘s disease activity index)。8項目の疾患活動性指数であり、3項目の患者報告項目と5項目の医師報告又は臨床検査項目の組合せで構成される。患者の7日間の症状を振り返って入力された電子日誌のデータを基にスコアを算出した。7日間の回答を合計し、すべての項目を重みづけした合計スコアは0~600ポイントとなり、スコアが高いほど重症であることを示す。 |
|---|
解析計画
有効性の解析はPAS集団注12)を、安全性の解析は安全性解析対象集団(mITT集団)注13)を対象として実施した。主要評価項目及び重要な副次評価項目では、全体での第1種の過誤の確率を両側有意水準0.05に制御するため、事前に規定したグラフィカルアプローチを実施した。グラフィカルアプローチを用いて検定する仮説は、主要評価項目及び重要な副次評価項目の仮説を含む2つのグループで構成された。また、特に指定しない限り、その他の副次評価項目(重要な副次評価項目を除く副次評価項目)では、名目上の有意水準を0.05として検定を実施し、名目上のp値を示すこととした。
グループ1:プラセボと比較する主要評価項目及びすべての重要な副次評価項目
グループ2:ウステキヌマブと比較するすべての重要な副次評価項目
グループ1では、全体の第1種の過誤の確率を0.005とした。グループ1の検定ですべての仮説が棄却された場合、グループ2の検定は全体の第1種の過誤の確率を0.05として実施した。グループ1の検定で主要評価項目の仮説が棄却され、残りの仮説が1つでも棄却されなかった場合、グループ2の検定は全体の第1種の過誤の確率を0.045として実施した。日本人集団の解析は、記述統計量の集計のみとした。主要評価項目などの二値変数の有効性評価項目の解析(プラセボ群又はウステキヌマブ群に対するオンボー®群の優越性の評価)では、Mantel-Haenszel推定量による信頼区間及びCMH検定を実施した。Mantel-Haenszel推定量による信頼区間及びCMH検定では、生物学的製剤不良の状態(あり/なし)、ベースラインのSES-CDスコア(12未満/12以上)、指示変数(ベースラインの排便回数が7回以上又はベースラインの腹痛スコアが2.5以上)を層別因子とした。
ウステキヌマブに対するオンボー®の非劣性を評価するため、重要な副次評価項目であるCDAIによる臨床的寛解を達成した患者の割合について、グラフィカルアプローチの結果に応じて用いる有意水準に基づき群間差の推定値の両側95%信頼区間又は95.5%信頼区間の下限値を非劣性マージンと比較した。重要な副次目的である52週のCDAIによる臨床的寛解では、非劣性マージンを10%とし、寛解達成の割合の群間差を用いた非劣性検定のp値も算出した。
連続量は、共分散分析(analysis of covariance:ANCOVA)を用いて解析した。ANCOVAでは、治験薬、生物学的製剤不良の状態(あり/なし)、ベースラインのSES- CDスコア(12未満/12以上)、指示変数(ベースラインの排便回数が7回以上又はベースラインの腹痛スコアが2.5以上)、ベースラインのスコアをモデルに含めた。
欠測データは、二値変数の評価項目の解析では複合ストラテジーを用いた。治験薬投与中止又はクローン病治療のための併用薬に特定の変更があった患者はノンレスポンダーとし、その時点以降のすべての来院はNRIを用いて補完した。Estimand注14)の観点から、これらの患者は欠測とみなさなかった。評価時点まで治験薬の投与を完了した患者のうち、二値の評価項目に散発的な欠測が認められた少数の患者には、NRIを用いて補完した。また、プラセボ群の患者が12週にミリキズマブ投与に変更した中間事象に対して仮想ストラテジーを用いた場合、12週以後のすべての来院はNRIを用いて補完した。連続量では、欠測値をmBOCFで補完したANCOVAを主要解析に用いた。
なお、有効性評価項目について、サブグループ解析注15)を行うことを事前に規定した。生物学的製剤の治療不良の有無別の群間比較はFisher’s exact検定を用いた。
その他の副次評価項目であるCDAIによる臨床的寛解を達成した患者の割合及び便意切迫感に関するNRSスコアのベースラインからの変化量の評価期間は、0週から52週の各来院時とした。
また、CDAI及びPROの構成要素である排便回数及び腹痛スコアの0週から52週の各来院時のベースラインからの変化量についても補足的に解析した。
- 注12)無作為割付され、1回以上の治験薬投与を受けた患者で、ベースライン時のSES-CDスコアが7以上(回腸限局型クローン病を有する患者では4以上)の患者。割付された治験薬を使用しなかった患者、正しい治験薬を使用しなかった患者、治験実施計画書を遵守しなかった患者も含めた。
- 注13)安全性解析対象集団はmITT集団と同じ集団。無作為割付され、1回以上の治験薬投与を受けたすべての患者。割付された治験薬を使用しなかった患者、正しい治験薬を使用しなかった患者、治験実施計画書を遵守しなかった患者も含めた。
- 注14)主要Estimandは、関心のある主要な臨床的疑問「PAS集団を対象に、クローン病治療のための併用薬に特定の変更がなく52週間の治験薬投与を遵守し、主要評価項目を達成した患者の割合それぞれについて、オンボー®とプラセボとの差はどの程度であるか。」に対応する。主要評価項目及び重要な副次評価項目のEstimandはPAS集団に適用し、集団レベルでの変数の要約として二値変数の評価項目では群間差を、連続変数の評価項目では最小二乗平均値の差を示した。
- 注15)以下の部分集団で解析を実施
- ・日本人
- ・生物学的製剤の使用歴なし、又は生物学的製剤で治療不良(効果不十分、効果減弱又は不耐)の経験がなかった患者(Not Biologic Failed)
- ・1種類以上の生物学的製剤で治療不良(効果不十分、効果減弱又は不耐)の経験があった患者(Biologic Failed)
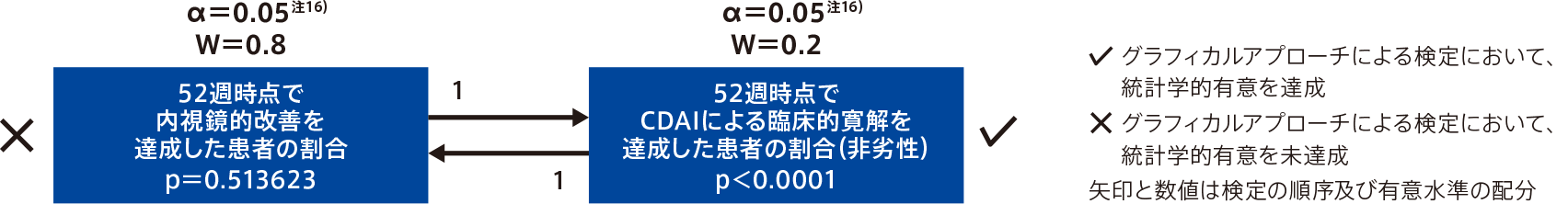
- 注16)グループ1の検定ですべての仮説が棄却されたため、グループ2の検定は全体の第1種の過誤の確率を0.05とした。