- 医療関係者向けホーム
- 精神科領域
- レクサプロ
- Clinical Study:第Ⅲ相パロキセチン対照二重盲検比較試験(海外データ)(うつ病・うつ状態) 試験の概要
 Clinical Study第Ⅲ相パロキセチン対照二重盲検比較試験(海外データ)(うつ病・うつ状態)
Clinical Study第Ⅲ相パロキセチン対照二重盲検比較試験(海外データ)(うつ病・うつ状態)
「禁忌を含む使用上の注意」等は添付文書をご参照ください。
試験の概要「第Ⅲ相パロキセチン対照二重盲検比較試験(海外データ)(うつ病・うつ状態)」
- Baldwin, D. S. et al.:Int Clin Psychopharmacol 21, 159-169(2006)
- 【試験実施体制:ルンドベック社の支援(出資、労務提供、共著者の一部)により実施】
試験デザイン
ランダム化、二重盲検並行群間比較試験
目的
大うつ病性障害患者を対象にエスシタロプラム10~20mg/日の有効性及び安全性について、パロキセチン20~40mg/日を対照として検討した。
対象
DSM-IVによる主診断が大うつ病性障害であり、MADRS合計点が22点以上40点以下の18歳以上の患者
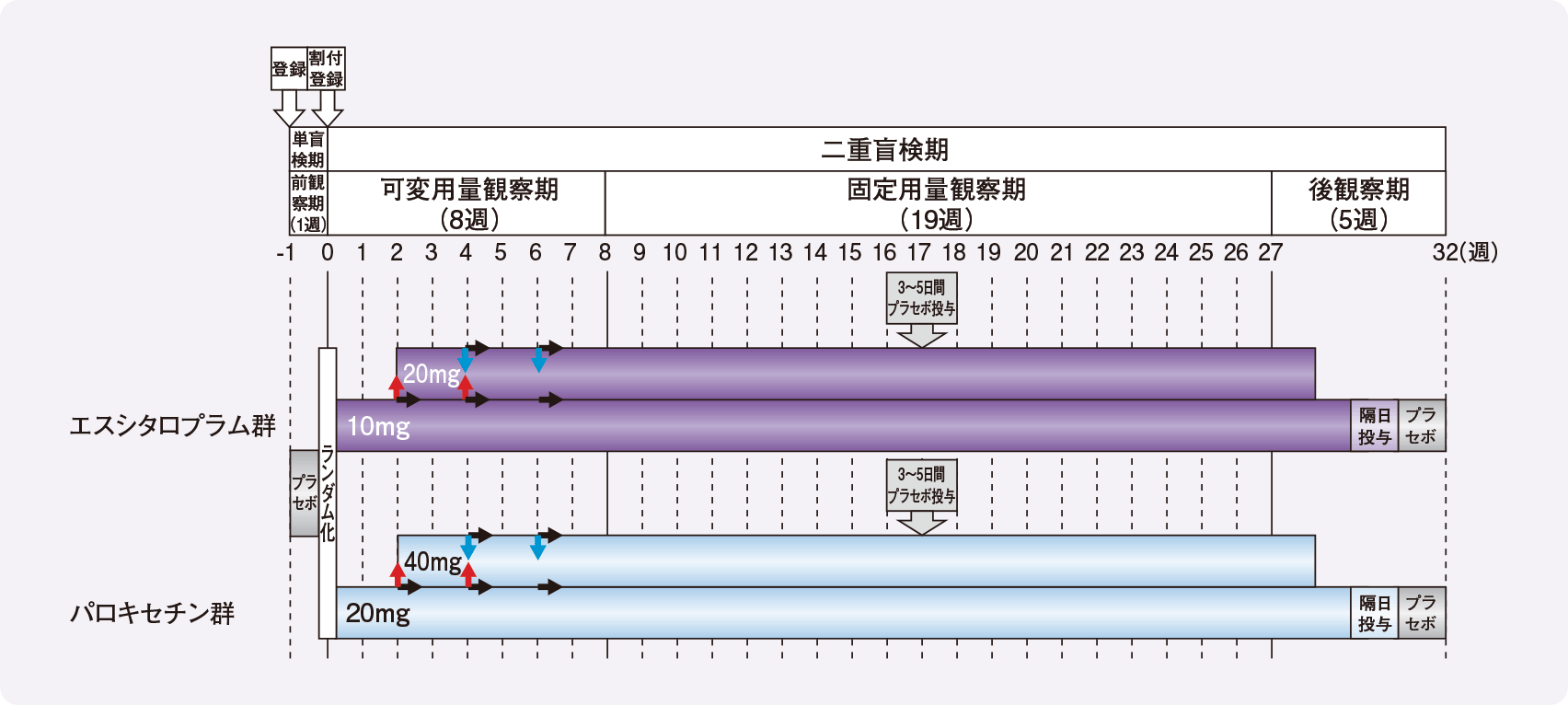
投与方法
- 前観察期:全例にプラセボを1週間投与
-
可変用量観察期:
エスシタロプラム群(165例): エスシタロプラム10mg/日を2週間投与後、2週もしくは4週時に20mg/日に増量可能、4週もしくは6週時に減量可能 パロキセチン群(158例): パロキセチン20mg/日を2週間投与後、2週もしくは4週時に40mg/日に増量可能、4週もしくは6週時に減量可能 - 固定用量観察期:両群とも、可変用量観察期8週時の用量を27週まで継続投与
ただし、コンプライアンス不良に伴う離脱症状を評価するため、固定用量観察期に8週間以上治療薬を投与した症例については、治療薬投与を3~5日間中断してプラセボを投与 - 後観察期:治療薬を1~3週間かけて漸減し、その後1週間隔日投与した後、プラセボを1~3週間投与
有効性評価項目
<主要評価項目>
MADRS合計点の変化量(27週時)
<副次評価項目>
27週時における以下の項目 ・MADRS合計点での寛解率(合計点が12点以下の患者の割合) ・MADRS合計点での反応率(合計点がベースラインより50%以上減少した患者の割合) ・ベースラインの重症度で層別化したサブグループにおけるMADRS合計点の変化量(30点以上又は30点未満)
安全性評価項目
有害事象発現率、投与中止率、DESSに基づく離脱症状、バイタルサイン(血圧、脈拍数)
解析計画
主要評価項目であるMADRS合計点の変化量については、ANCOVAを用いて投与群及び試験施設を因子、ベースラインのMADRS合計点を共変量として、投与群間の比較を行う。
(注意)パロキセチンの「うつ病・うつ状態」に対して承認されている用法及び用量は次の通りです。
通常、成人には1日1回夕食後、パロキセチンとして20~40mgを経口投与する。投与は1回10~20mgより開始し、原則として1週ごとに10mg/日ずつ増量する。なお、症状により1日40mgを超えない範囲で適宜増減する。

