- 医療関係者向けホーム
- 精神科領域
- レクサプロ
- Clinical Study:第Ⅲ相プラセボ・パロキセチン対照二重盲検比較試験(用量反応非劣性試験)(うつ病・うつ状態) 試験の概要
 Clinical Study第Ⅲ相プラセボ・パロキセチン対照二重盲検比較試験(用量反応非劣性試験)(うつ病・うつ状態)
Clinical Study第Ⅲ相プラセボ・パロキセチン対照二重盲検比較試験(用量反応非劣性試験)(うつ病・うつ状態)
「禁忌を含む使用上の注意」等は添付文書をご参照ください。
試験の概要「第Ⅲ相プラセボ・パロキセチン対照二重盲検比較試験(用量反応非劣性試験)(うつ病・うつ状態)」
- 持田製薬社内資料 : 用量反応非劣性試験-大うつ病性障害患者におけるプラセボ及び塩酸パロキセチンを対照とした有効性及び安全性の検討-
(2011年4月22日承認、CTD 2.7.6.8.2)<承認時評価資料>
試験デザイン
ランダム化、二重盲検並行群間比較試験
目的
大うつ病性障害患者を対象にエスシタロプラム10mg/日及び20mg/日の有効性(プラセボに対する優越性、パロキセチン20~40mg/日に対する非劣性;MADRS合計点の変化量)と安全性を検討した。
対象
DSM-IV-TRによる主診断が大うつ病性障害であり、MADRS合計点が22点以上かつCGI-Sが4点以上を満たす20歳以上65歳未満の外来患者
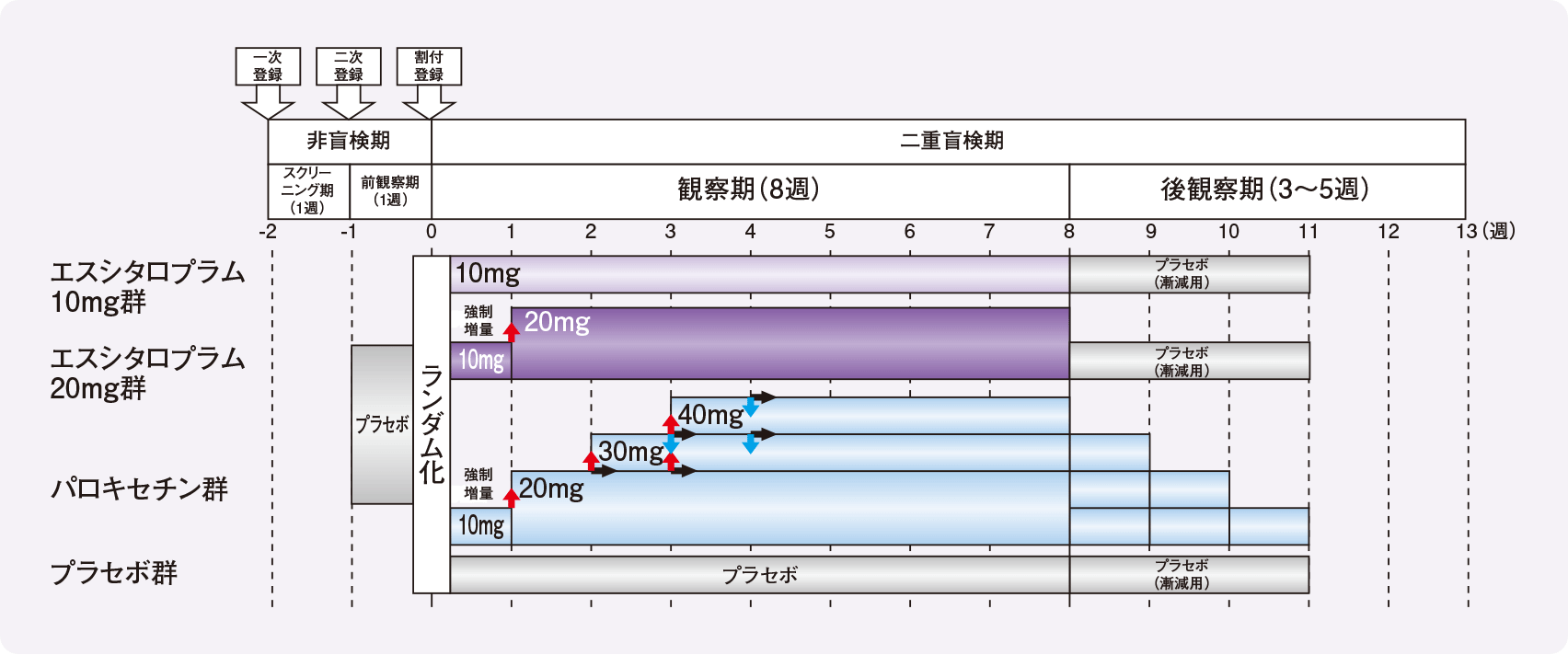
投与方法
- 前観察期:全例にプラセボを1週間投与
-
観察期:
- エスシタロプラム10mg群(120例)※:エスシタロプラム10mg/日を8週間投与
- エスシタロプラム20mg群(119例)※:エスシタロプラム10mg/日を1週間投与後、20mg/日を7週間投与
- パロキセチン群(121例):パロキセチン10mg/日を1週間投与後、20mg/日を1週間投与し、2週、3週、4週時に10mg/日ずつ増減(最低用量20mg/日、最高用量40mg/日)して計8週間投与
- プラセボ群(124例):プラセボを8週間投与
-
後観察期:
- エスシタロプラム10mg群、20mg群及びプラセボ群:漸減用プラセボを1~3週間投与
- パロキセチン群:1週ごとに10mg/日ずつ漸減
- ※:エスシタロプラム10mg群と20mg群を合わせた群をエスシタロプラム併合群とした。
有効性評価項目
<主要評価項目>
MADRS合計点の変化量 (8週時)
<副次評価項目>
8週時における以下の項目
・MADRS合計点での反応率(合計点が治療開始時から50%以上減少した患者の割合) ・HAM-D17合計点の変化量 ・HAM-D17合計点での反応率(合計点が治療開始時から50%以上減少した患者の割合)
<その他の評価項目>
8週時における以下の項目
・MADRS合計点での寛解率(合計点が10点以下の患者の割合)・HAM-D17合計点での寛解率(合計点が7点以下の患者の割合) ・HAM-Dサブスケール(抑うつ気分・身体についての不安・精神運動抑制・睡眠障害・認知障害・憂うつ症状)の変化量 ・CGI-Sの変化量 ・CGI-I
安全性評価項目
副作用発現率、一般臨床検査(血液学的検査、血液生化学検査、尿検査)、バイタルサイン(血圧、脈拍数)及び体重、心電図
解析計画
主要評価項目であるMADRS合計点の変化について、エスシタロプラム群のプラセボ群に対する優越性の検証及びパロキセチン群に対する非劣性の検証にはいずれもベースラインのMADRS合計点を共変量としたANCOVAを用い、パロキセチン群に対する非劣性の検証についてはさらに2群間のMADRS合計点の変化量の平均値の差の両側95%信頼区間を用いて評価する。

