- 医療関係者向けホーム
- 消化器領域
- リアルダ®錠
- Clinical Study:成人(各臨床試験での定義:16歳以上)の潰瘍性大腸炎患者に対する臨床成績:寛解期 試験の概要
 Clinical Study
Clinical Study
成人(各臨床試験での定義:16歳以上)の潰瘍性大腸炎患者に対する臨床成績:寛解期
寛解期:リアルダ1200mg錠と600mg錠の治療学的同等性
リアルダ1200mg錠から600mg錠への切り替えを検討した寛解期における国内第Ⅲ相試験
-
[持田製薬社内資料:国内第Ⅲ相試験 ―寛解期の潰瘍性大腸炎を対象とした、
1200mg錠から600mg錠への切替えにおける有効性及び安全性の検討―
(2025年6月24日承認、CTD 2.7.6.6)]〈承認時評価資料〉
試験の概要
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
試験デザイン
多施設共同・非盲検・非対照試験
目的
リアルダ1200mg錠注)と600mg錠注)の治療学的同等性を評価するため、寛解期の潰瘍性大腸炎に対するリアルダ1200mg錠から600mg錠への切り替えにおける有効性および安全性を検討することを目的とした。
対象
寛解期の潰瘍性大腸炎患者24例
〔評価期1におけるFAS(Full Analysis Set):23例、評価期2におけるFAS:23例、評価期1における安全性解析対象集団:23例、評価期2における安全性解析対象集団:23例〕
| 【選択基準】 |
|
|---|
- ※1:UC-DAIスコアを構成する4つの評価項目〔排便回数、血便、S状結腸の粘膜所見、PGA(医師による全般的評価)〕のうち、S状結腸の粘膜所見を除く3つの項目のこと。
- ※2:寛解の定義は、治験責任(分担)医師により、潰瘍性大腸炎に由来すると判断される血便が認められていない状態とする。
方法
試験期間は、前観察期(2週間)、投与期〔16週間(評価期1:8週間、評価期2:8週間)〕、後観察期(1週間)から構成された。16週間にわたる投与期のうち、評価期1(8週間)ではリアルダ1200mg錠を1日1回2錠、朝食後に8週間経口投与し、評価期2(8週間)ではリアルダ600mg錠を1日1回4錠、朝食後に8週間経口投与した。
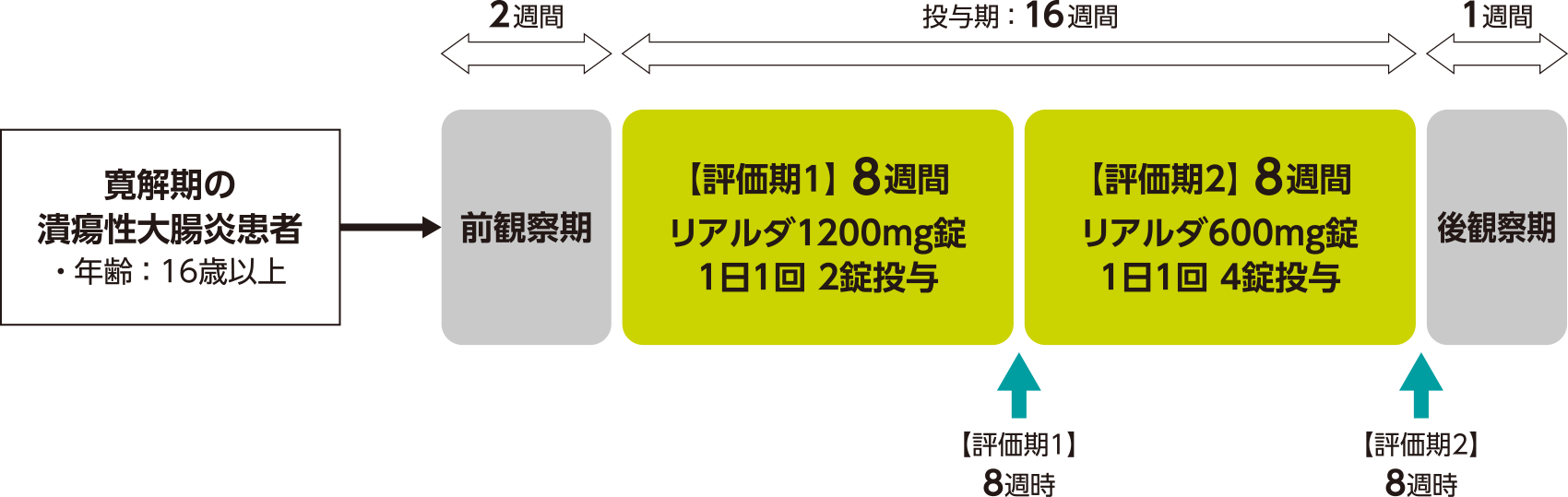
主要評価項目
| 〈有効性〉 | 血便の非発現率[評価期1、評価期2](該当期のいずれの来院時にも血便スコア0) |
|---|---|
| 〈安全性〉 | 有害事象[評価期1、評価期2] |
副次評価項目
| 〈有効性〉 | 血便の非発現[投与期](投与期のいずれの来院時にも血便スコア0)、部分UC-DAIスコアの該当期開始時からの変化量[評価期1、評価期2、投与期] など |
|---|---|
| 〈安全性〉 | 有害事象[投与期]、副作用[評価期1、評価期2、投与期] |
UC-DAIスコアの概要については、こちらをご参照ください。
解析計画
有効性の主要な解析対象集団は、評価期1および評価期2におけるFASとした。安全性の主要な解析対象集団は、評価期1および評価期2における安全性解析対象集団とした。
主要評価項目の「血便の非発現」については、該当期のいずれの来院時にも血便スコアが0(血便の非発現)であった患者の割合(血便の非発現率)およびその両側95%信頼区間を算出した。評価期1と評価期2の「血便の非発現率」の差(評価期2-評価期1)が「±12%以内」の場合、リアルダ1200mg錠と600mg錠の有効性は同等であると判断することとした。
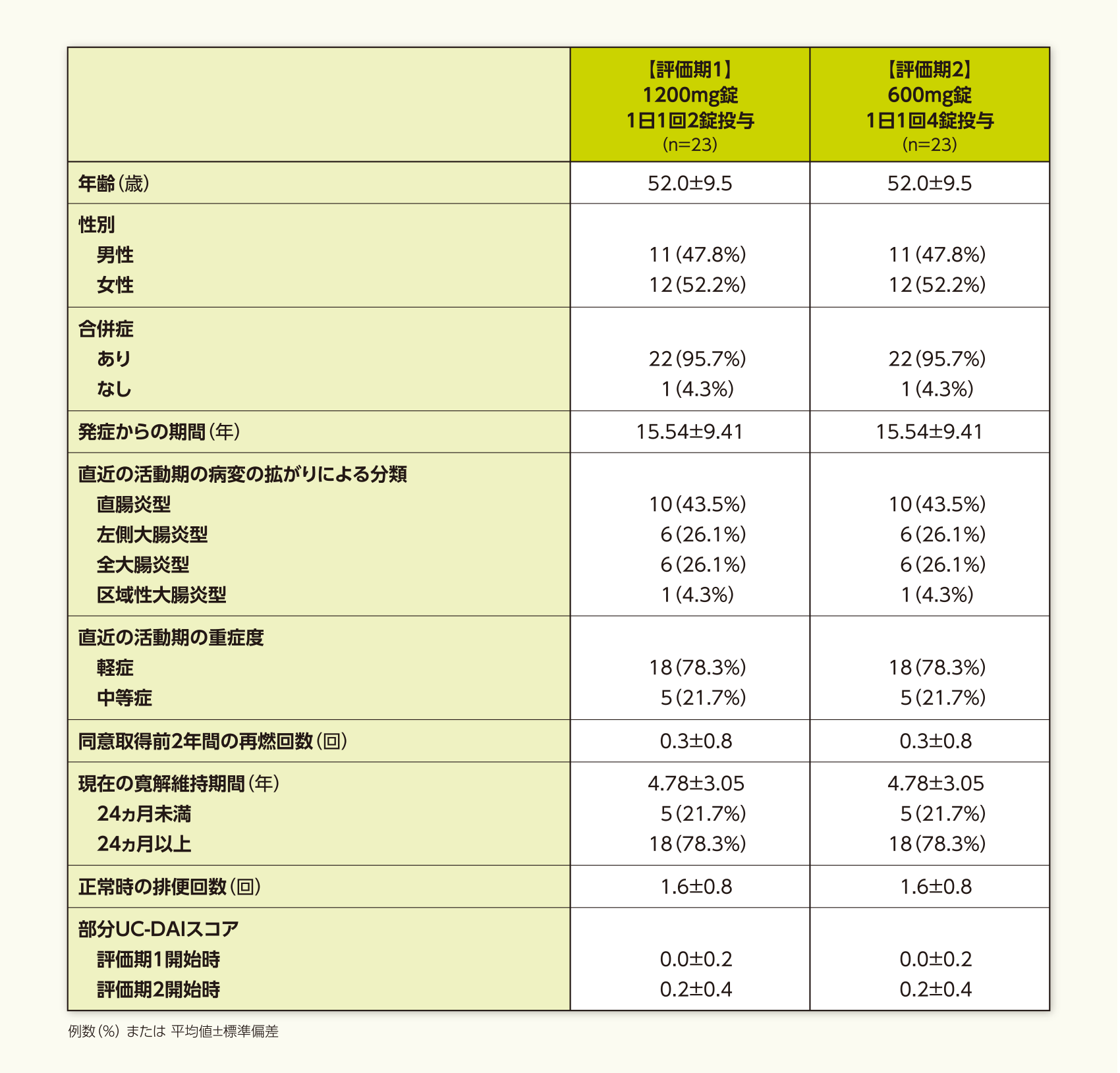
患者背景(FAS)
- 注)1200mg錠と600mg錠との生物学的同等性を評価する試験は実施されていません。
- 7. 用法及び用量に関連する注意
- (抜粋)
- 7.4 使用製剤(本剤1200mg錠及び600mg錠)を切替える場合は、患者の状態を慎重に観察すること。[17.1.3 参照]
2025年12月作成
14407-18/N14 B8 MDC

