- 医療関係者向けホーム
- 消化器領域
- モビコール®配合内用剤
- Clinical Study:成人国内第Ⅲ相試験(検証期:プラセボ対照試験) 試験の概要
 Clinical Study成人国内第Ⅲ相試験(検証期:プラセボ対照試験)
Clinical Study成人国内第Ⅲ相試験(検証期:プラセボ対照試験)
「モビコール」及びMOVICOLは、Norgineグループの登録商標です。
試験の概要「成人国内第Ⅲ相試験(検証期:プラセボ対照試験)1)」
1)EAファーマ株式会社:社内資料(成人国内第Ⅲ相試験)<承認時評価資料>
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
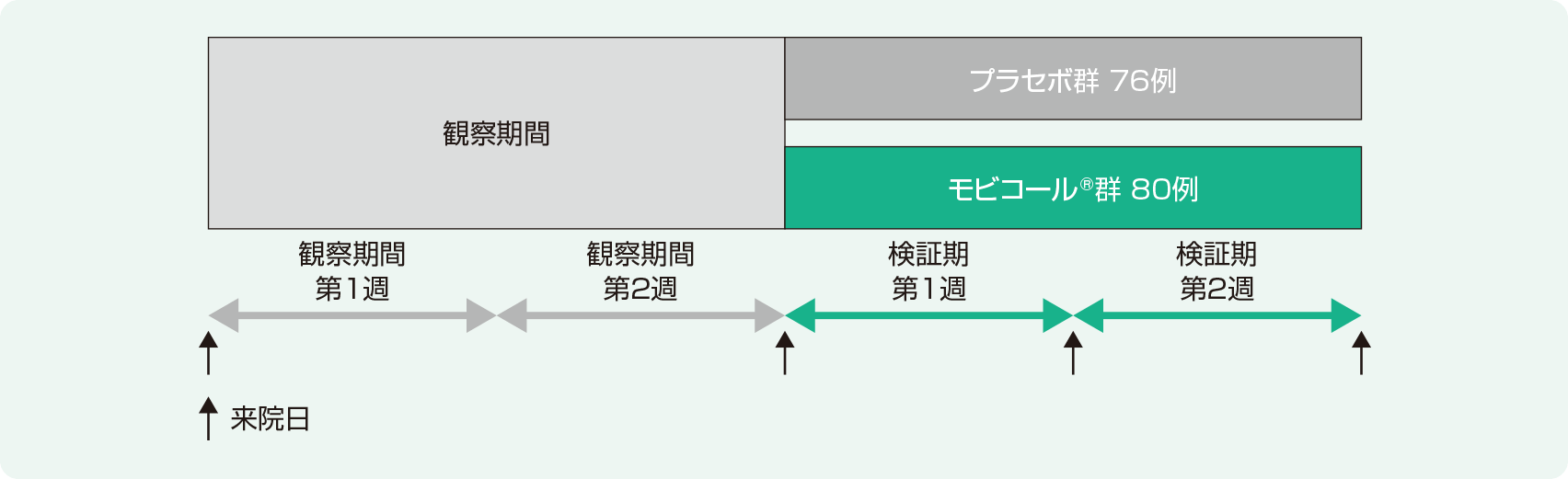
試験デザイン
プラセボ対照無作為化二重盲検多施設共同並行群間比較試験
目的
慢性便秘症患者を対象にモビコール®を2週間経口投与したときの有効性を検証するとともに、安全性を検討する。
対象
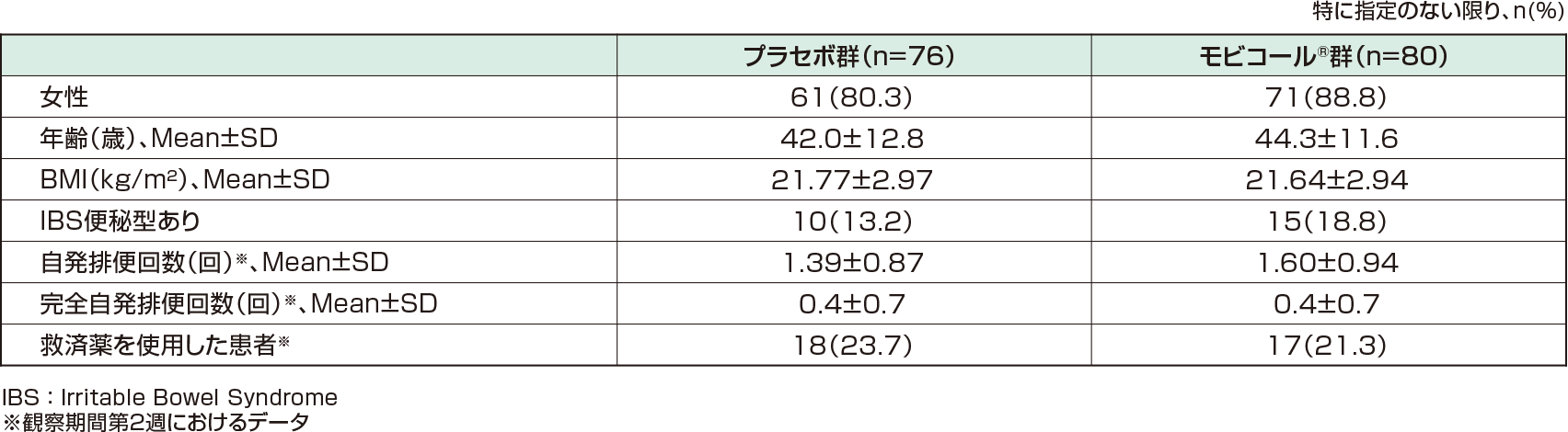
以下の条件を満たす15歳以上の慢性便秘症患者156例(有効性解析対象[FAS※1]:156例、安全性解析対象:156例)(プラセボ群76例、モビコール®群80例)。
- 同意取得時の6ヵ月以上前から自発排便回数が平均3回/週未満である。
- 同意取得時の6ヵ月以上前から自発排便に関連した以下の①~③の症状を1つ以上有している。
①排便の25%以上にいきみがある。
②排便の25%以上に兎糞状便または硬便がある。
③排便の25%以上に残便感がある。 - 20歳以上(同意取得時)の患者の場合、5年以内に実施された大腸内視鏡検査または注腸X線造影検査にて、大腸内に器質的に問題となる病変のないことが確認された患者
- 2週間の観察期間の自発排便回数が6回未満の患者 等
- ※1FAS:Full Analysis Set(最大の解析対象集団)モビコール®またはプラセボが1回以上投与され、何らかの有効性に関する観測値を持つすべての患者による集団を「最大の解析対象集団」とした。
投与方法
モビコール®6.9g包※2及びプラセボ6.9g包は、1包あたり約62.5mLの水に溶解した。
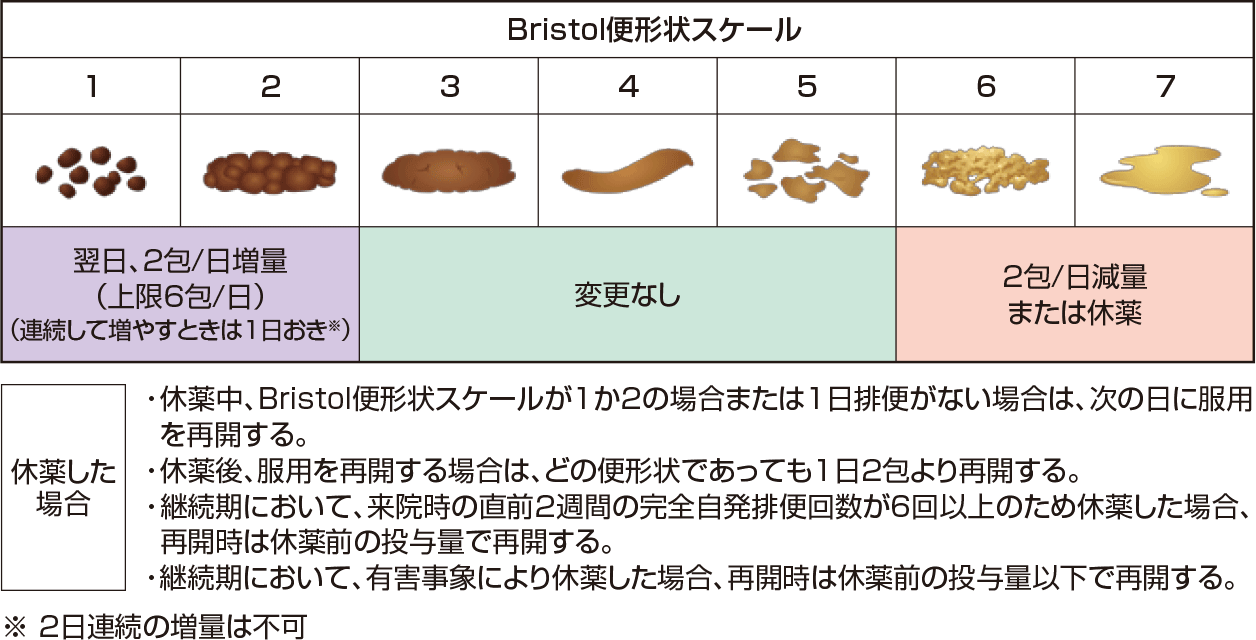
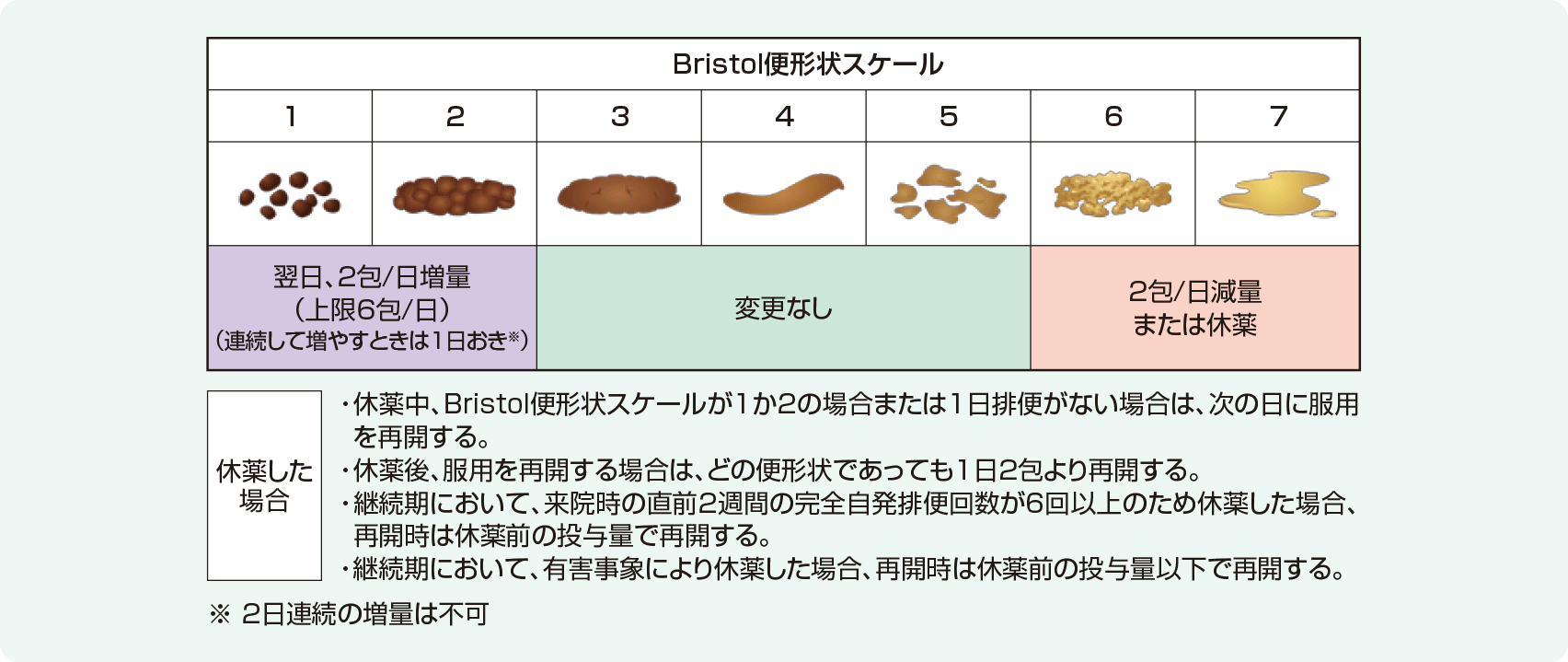
2週間の観察期間の後、モビコール®またはプラセボを2週間経口投与した。開始用量は1日1回2包とし、患者の状態により投与量調節基準を目安に投与量を調整した。なお、増量する場合は1日おきに行い、投与量の上限は1日6包とした。投与のタイミングは投与量に応じて1日1回または2回とし、1日2回の場合は朝、夕に投与した。仮登録から最終の観察・検査まで、連続して72時間以上排便が認められない場合に限り、ビサコジル坐剤10mgを救済薬として使用することを可とした。救済薬を1回使用しても排便が認められない場合には、試験継続の可否を医師が判断した。ただし、モビコール®またはプラセボ投与開始日前日および投与開始2日間の救済薬の使用は禁止した。
【用法及び用量】 -一部抜粋-(12歳以上の小児及び成人の用量のみ記載)
- ※2モビコール®6.9g包(有効成分:6.8523g):「モビコール®6.9g包」はモビコール®配合内用剤LD 1包(6.8523g)に相当する。
評価項目
有効性に関する評価項目※3
<主要評価項目(検証的評価項目)>
- 検証期第2週の自発排便回数の観察期間第2週からの変化量
<副次評価項目>
- 検証期第2週の完全自発排便回数の観察期間第2週からの変化量
- 検証期第1週および第2週の自発排便回数および完全自発排便回数におけるレスポンダーの割合
- 初回自発排便発現までの日数
- 初回完全自発排便発現までの日数
- Bristol便形状スケールに基づいた便硬度 等
- ※3有効性評価は患者日誌のデータに基づき行われた。
安全性に関する評価項目
- 有害事象
解析計画
主要評価項目(検証的評価項目)の主解析は、「検証期第2週の自発排便回数の観察期間第2週からの変化量※4」に正規分布を仮定し、観察期間第2週の自発排便回数を共変量とした共分散分析(ANCOVA)を適用した。解析結果として共分散分析で調整された「自発排便回数の変化量の差」の推定値、95%信頼区間、p値を算出した。
副次評価項目である検証期第2週の完全自発排便回数の観察期間第2週からの変化量※4は、各被験者における完全自発排便回数の変化量を求め、投与群ごとに要約統計量および平均値の95%信頼区間を求めた。
検証期第1週および第2週の自発排便回数および完全自発排便回数におけるレスポンダーの割合は、投与群ごとにレスポンダーの割合とその95%信頼区間を算出した。χ2検定あるいはFisherの直接確率法による検定を行った。初回自発排便発現までの日数および初回完全自発排便発現までの日数は、投与群ごとにKaplan-Meier法による中央値の算出、およびLog-rank検定を行った。
Bristol便形状スケールに基づいた便硬度は、便硬度の平均値を連続量として取扱い、投与群ごとに要約統計量を算出した。また、評価期間内の便硬度の中央値を算出して各スコアの頻度表を作成し、各週において群間比較をWilcoxon順位和検定で行った。
安全性の解析は検証期を対象に、投与群別に実施した。安全性解析対象集団はモビコール®またはプラセボが1回以上投与された患者とした。
- ※4変化量=観察期間第2週データをベースライン値とした。
- 検証的な解析以外で得られたp値を名目上のp値、95%信頼区間を探索的結果とした。

用語の定義
| 自発排便 | 下剤/浣腸または摘便なしに発現する排便。本試験においては、救済薬※使用後24時間以内の排便は自発排便としない。 |
|---|---|
| 完全自発排便 | 残便感のない自発排便。 |
| レスポンダー | 1週間あたりの自発排便回数または完全自発排便回数が、観察期間第2週より1回以上改善し、かつ3回以上である患者。 |
- ※成人国内第Ⅲ相試験においては、連続して72時間以上排便が認められない場合に限り、ビサコジル坐剤10mgを救済薬として使用することを可とした。
6. 用法及び用量 –一部抜粋-(12歳以上の小児及び成人の用量のみ記載)
本剤は、水で溶解して経口投与する。
通常、成人及び12歳以上の小児には初回用量としてLD 2包又はHD 1包を1日1回経口投与する。以降、症状に応じて適宜増減し、1日1~3回経口投与、最大投与量は1日量としてLD 6包又はHD 3包まで(1回量としてLD 4包又はHD 2包まで)とする。ただし、増量は2日以上の間隔をあけて行い、増量幅は1日量としてLD 2包又はHD 1包までとする。
ビサコジル坐剤の使用にあたっては、電子添文をご確認ください。