- 医療関係者向けホーム
- 消化器領域
- オンボー®
- Clinical Study(クローン病)「クローン病」に対する効果 日本人部分集団(サブグループ解析)
 Clinical Study(クローン病)「クローン病」に対する効果
Clinical Study(クローン病)「クローン病」に対する効果
日本人部分集団(サブグループ解析)
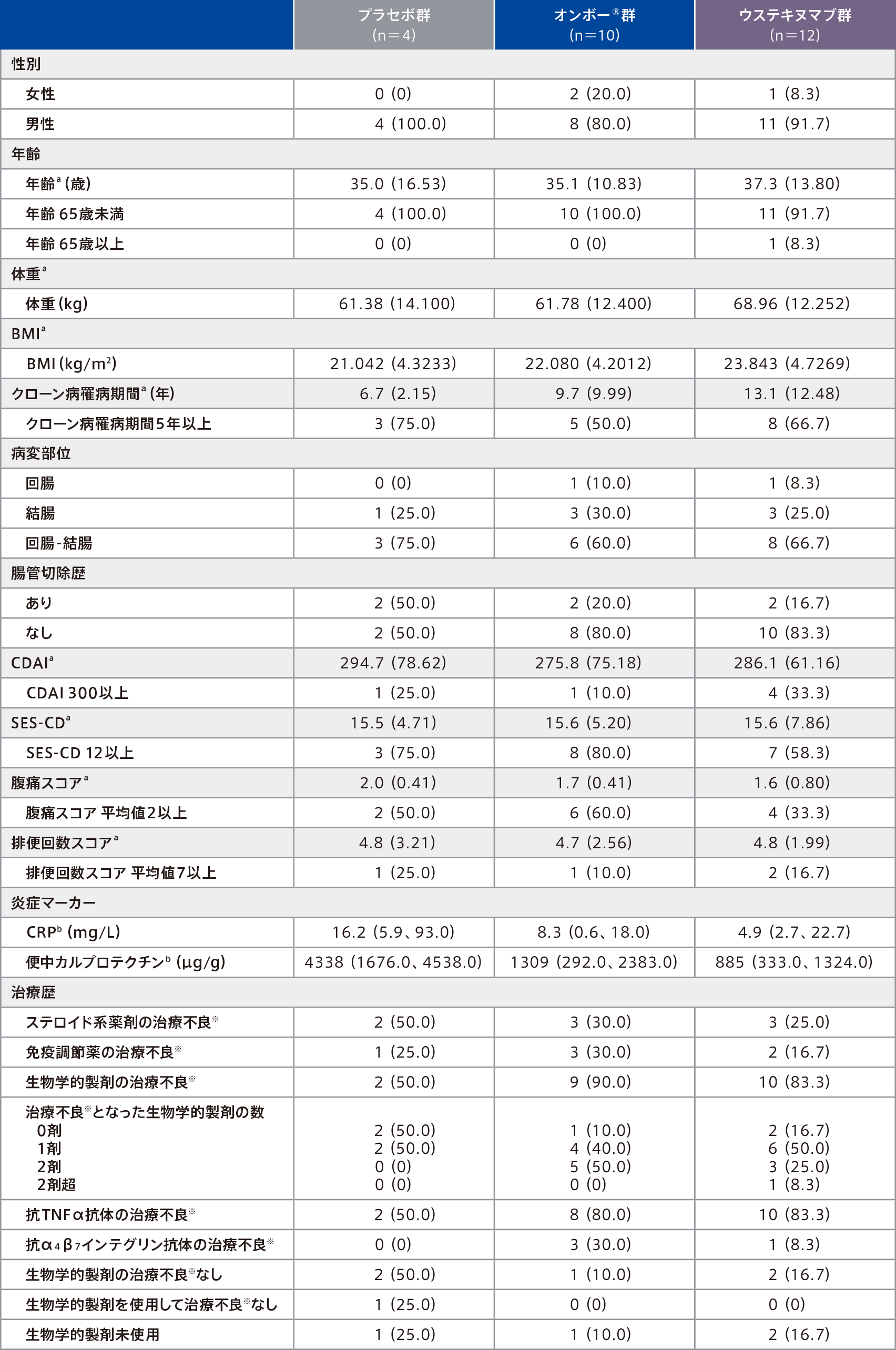
患者背景
有効性(プラセボ群との比較)
●12週時点でPROによる臨床的改善かつ52週時点で内視鏡的改善を達成した患者の割合
(主要評価項目のサブグループ解析:日本人部分集団)
●12週時点でPROによる臨床的改善かつ52週時点でCDAIによる臨床的寛解を達成した患者の割合
(主要評価項目のサブグループ解析:日本人部分集団)
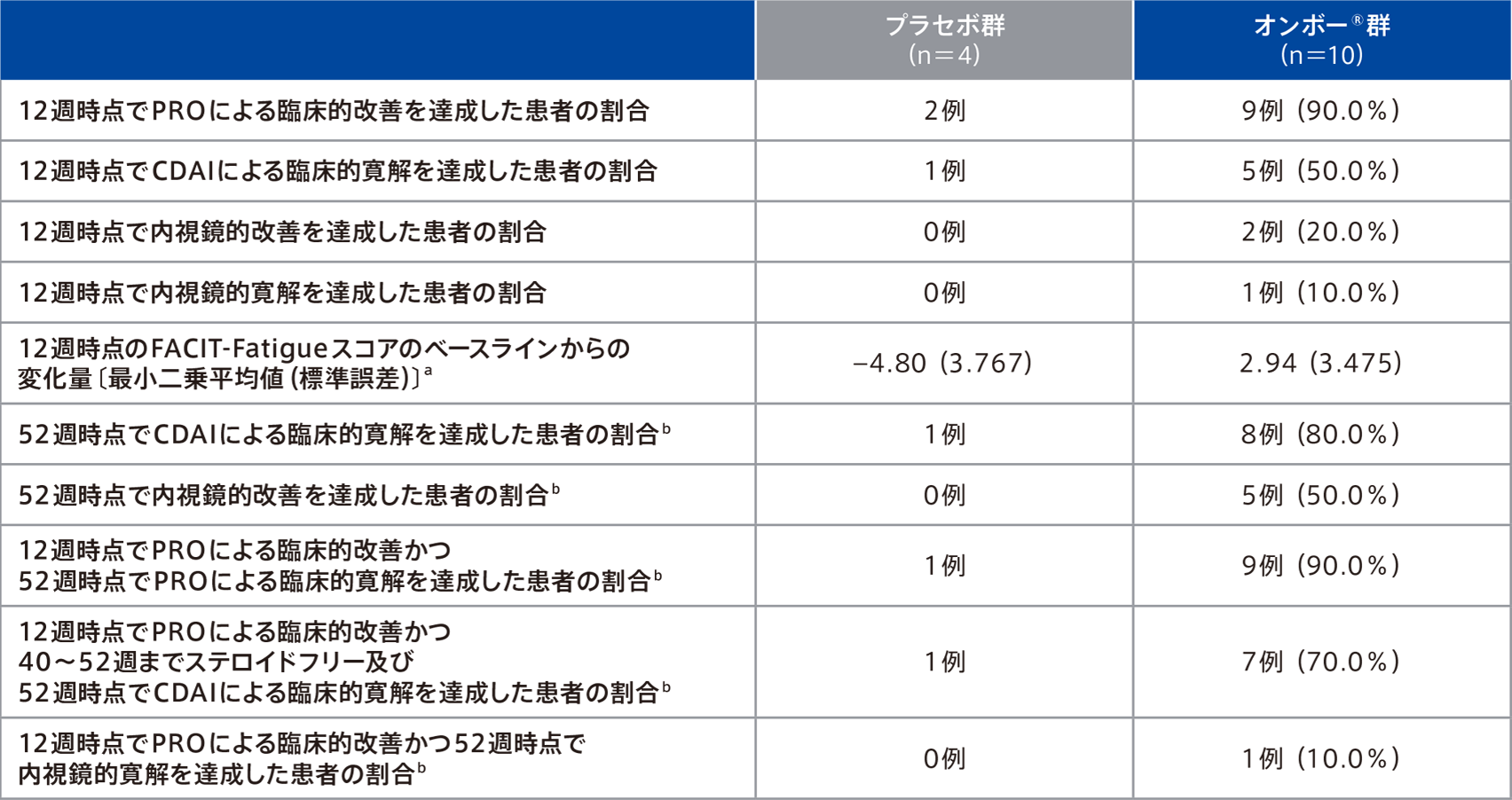
日本人部分集団における12週時点でPROによる臨床的改善かつ52週時点で内視鏡的改善を達成した患者の割合は、オンボー®群40.0%(4/10例)、プラセボ群4例中0例であった。
日本人部分集団における12週時点でPROによる臨床的改善かつ52週時点で臨床的寛解を達成した患者の割合は、オンボー®群70.0%(7/10例)、プラセボ群4例中1例であった。

PAS 、NRI
- プラセボ群に割り付けられた12週時点のノンレスポンダーでオンボー®投与に移行した患者はオンボー®群に含まない
- プラセボ群に割り付けられ12週時点でノンレスポンダーと判定された患者は、12週以降はプラセボ群のノンレスポンダーとして扱われた

●重要な副次評価項目のサブグループ解析:日本人部分集団
日本人部分集団における重要な副次評価項目の結果は下表のとおりであった。
PAS、NRI(12週時点のFACIT-Fatigueスコアのベースラインからの変化量以外)
- a PAS 、mBOCF
- b プラセボ群に割り付けられた12週時点のノンレスポンダーでオンボー®投与に移行した患者はオンボー®群に含まない
プラセボ群に割り付けられ12週時点でノンレスポンダーと判定された患者は、12週以降はプラセボ群のノンレスポンダーとして扱われた
評価項目の用語の定義についてはVIVID-1試験の試験概要参照
有効性(ウステキヌマブ群との比較)
●52週時点でCDAIによる臨床的寛解を達成した患者の割合
(重要な副次評価項目のサブグループ解析:日本人部分集団)
●52週時点で内視鏡的改善を達成した患者の割合
(重要な副次評価項目のサブグループ解析:日本人部分集団)
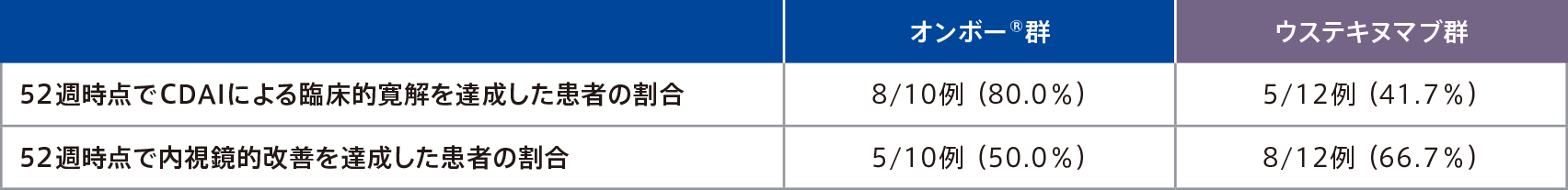
日本人部分集団における52週時点でCDAIによる臨床的寛解を達成した患者の割合は、オンボー®群80.0%(8/10例)、ウステキヌマブ群41.7%(5/12例)であった。
日本人部分集団における52週時点で内視鏡的改善を達成した患者の割合は、オンボー®群50.0%(5/10例)、ウステキヌマブ群66. 7%(8/12例)であった。
PAS 、NRI
- プラセボ群に割り付けられた12週時点のノンレスポンダーでオンボー®投与に移行した患者はオンボー®群に含まない


ウステキヌマブ皮下注のクローン病に対しての国内で承認された用法及び用量
ウステキヌマブ(遺伝子組換え)の点滴静注製剤を投与8週後に、通常、成人にはウステキヌマブ(遺伝子組換え)として90mgを皮下投与し、以降は12週間隔で90mgを皮下投与する。なお、効果が減弱した場合には、投与間隔を8週間に短縮できる。
安全性
●サブグループ解析:日本人部分集団
<第1期(0~12週)>
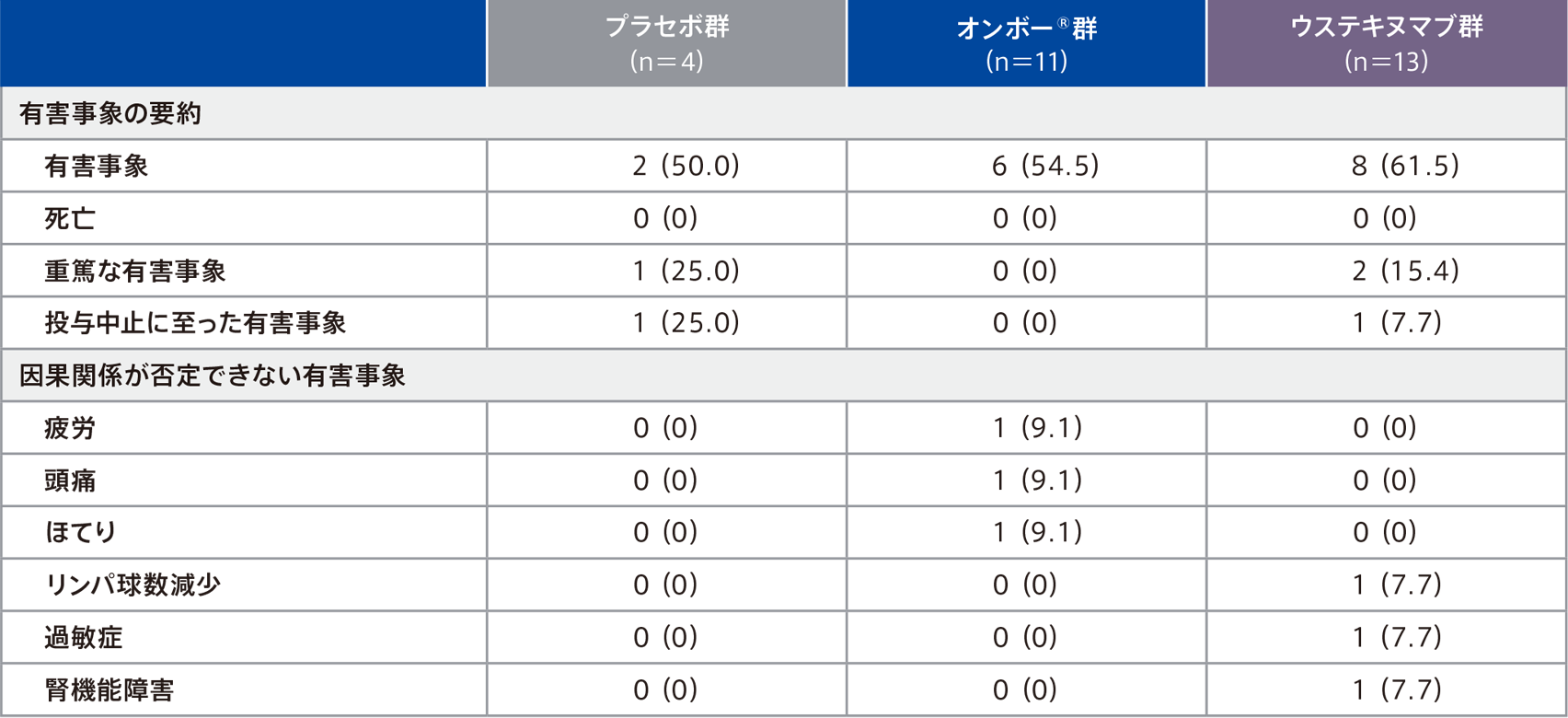
第1期での有害事象は、プラセボ群50.0%(2/4例)、オンボー®群54.5%(6/11例)、ウステキヌマブ群61.5%(8/13例)に認められ、主な有害事象(いずれかの群で2例以上に認められた有害事象)はオンボー®群の嘔吐2例(18.2%)であった。
因果関係が否定できない有害事象は、プラセボ群0%(0/4例)、オンボー®群18. 2%(2/11例)、ウステキヌマブ群23.1%(3/13例)に認められ、その事象は下表のとおりであった。
本試験において、日本人患者の死亡は認められなかった。
重篤な有害事象は、プラセボ群25.0%(1/4例、クローン病1例)、オンボー®群0%(0/11例)、ウステキヌマブ群15.4%(2/13例、肛門直腸障害、脱水各1例)に認められ、治験担当医師により治験薬との因果関係はないと判断された。
投与中止に至った有害事象は、プラセボ群1例(クローン病)、ウステキヌマブ群1例(肛門直腸障害)に認められた。
例数(%)、安全性解析対象集団(mITT集団)※
- ※安全性解析対象集団はmITT集団と同じ集団。無作為割付され、1回以上の治験薬投与を受けたすべての患者。割付された治験薬を使用しなかった患者、正しい治験薬を使用しなかった患者、治験実施計画書を遵守しなかった患者を含む。
<投与期全体>
投与期全体での有害事象は、プラセボ群100%(4/4例)、オンボー® 群81. 8%(9/11例)、ウステキヌマブ群100%(13/13例)に認められ、主な有害事象(いずれかの群で2例以上に認められた有害事象)は下表のとおりであった。
因果関係が否定できない有害事象は、プラセボ群0%(0/4 例)、オンボー®群27.3%(3/11例)、ウステキヌマブ群30.8%(4/13例)に認められ、オンボー® 群では疲労、頭痛、貧血、ほてり各1例(9.1%)、ウステキヌマブ群ではクローン病、湿疹、リンパ球数減少、過敏症、腎機能障害各1例(7.7%)であった。
本試験において、日本人患者の死亡は認められなかった。
重篤な有害事象は、プラセボ群25.0%(1/4例、クローン病1例)、オンボー®群9.1%(1/11例、貧血1例)、ウステキヌマブ群30.8%(4/13例、クローン病、肛門直腸障害、COVID-19、脱水各1例)に認められ、このうち、オンボー® 群の貧血1例、ウステキヌマブ群のクローン病1例は、治験薬との因果関係が否定できないと判断された。
投与中止に至った有害事象は、プラセボ群1例(クローン病)、ウステキヌマブ群2例(クローン病、肛門直腸障害各1例)に認められた。
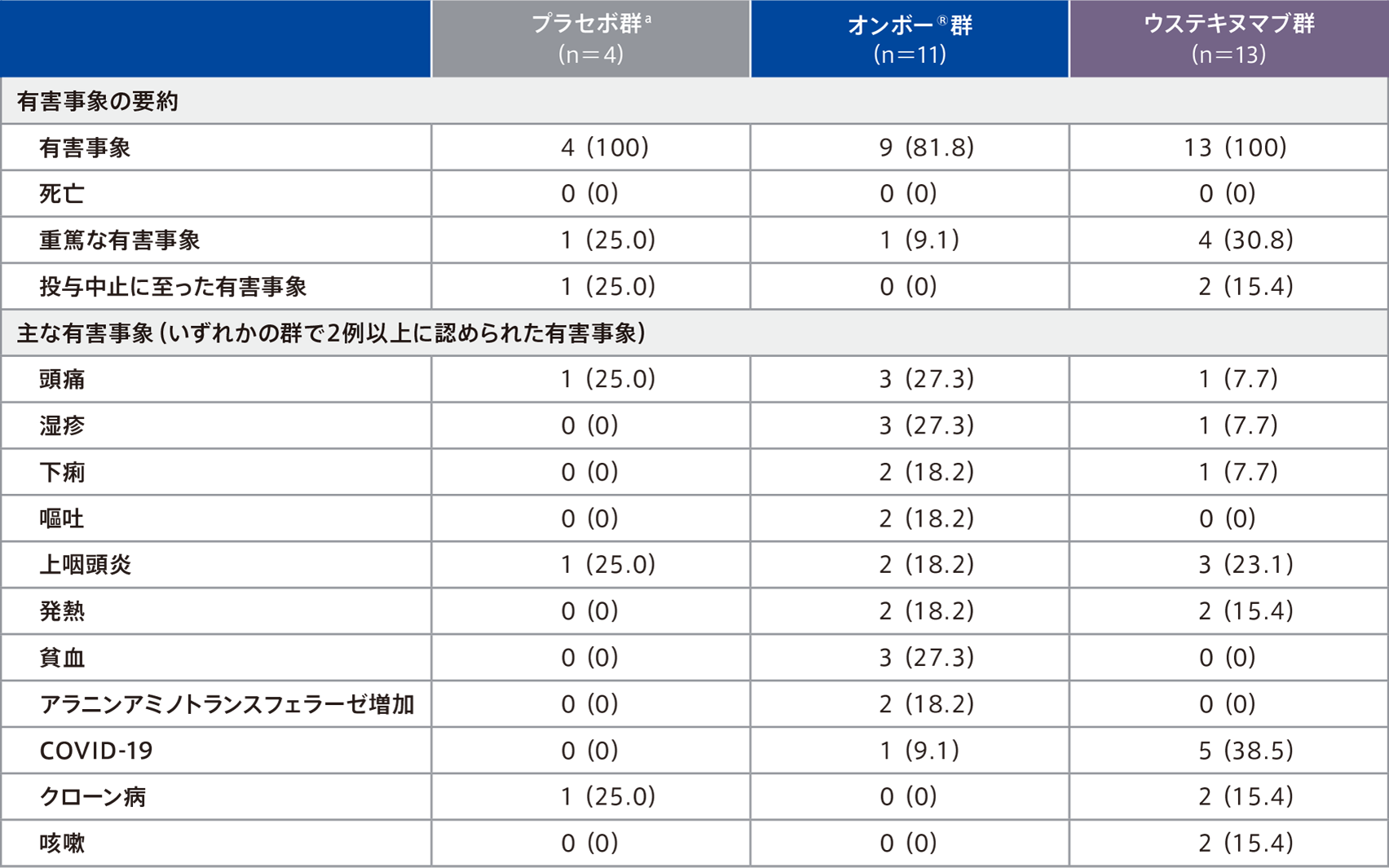
例数(%)、安全性解析対象集団(mITT集団)※
- ※安全性解析対象集団はmITT集団と同じ集団。無作為割付され、1回以上の治験薬投与を受けたすべての患者。割付された治験薬を使用しなかった患者、正しい治験薬を使用しなかった患者、治験実施計画書を遵守しなかった患者を含む。